Hi,
I just wanted to ask about when fold change and P-values are calculated.
If i wanted to do normalisation or scaling, would this then be reflected in the fold change calculations?
If fold change is calculated prior to any normalisation, is that standard acceptable practice? Or should i normalise it first myself, and then put it into metaboanalyst.
I am working with proteomics data so a conventional approach would be to log2 transform and then groupA-groupB to calculate fold change.
Any feedback is appreciated.
Yes, normalization and scaling are conducted before the fold-change and p-values are calculated, so they will impact these values. This is standard practice. You can see based on the order of the pages, and also you can click to see the R commands in the top right corner. This shows the order that different functions are called in.
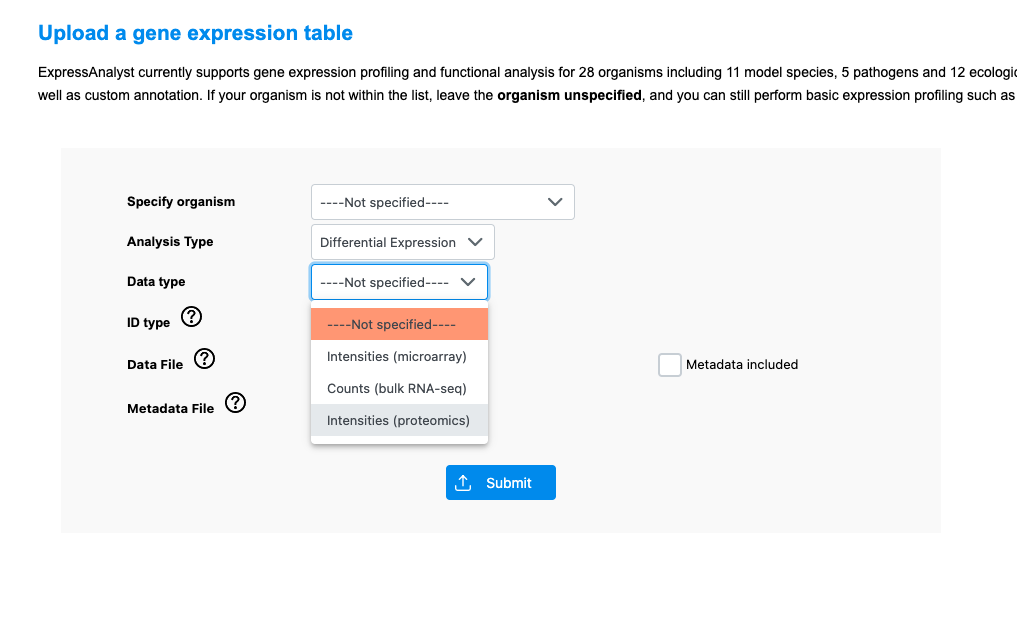
However, if you are analyzing proteomics data, I suggest using ExpressAnalyst. It is a tool designed for gene expression analysis, also by our lab and using the same general design as MetaboAnalyst. We recently added a “proteomics” data input type, and have added proteomics-specific normalization methods. This way you can conduct pathway analysis on the results after, and also ExpressAnalyst accepts proteomics-specific IDs like Uniprot.
Go to the ‘single expression table’ module, then select the proteomics data input type:
Hi Really useful thank you.
So when i apply log10 transformation to my data on metaboanalyst and compare it to the normal data the log2FC values don’t change? However when i apply log2(x) on express analyst i see a different fold change between transformed vs non-transformed (which is expected). The same applies on metaboanalyst for something like pareto scaling, there is no difference in fold change values between scaled and raw data.
Anyway express analyst gives me the results i would expect, but just interested in why i might be seeing no changes with scaling and transformation.
Thanks for your help
The sentences below are copy-and-pasted directly from the fold analysis page:
Fold Change (FC) Analysis
The goal of fold change analysis is to compare the absolute values of change between two group means. Since column-wise normalization (i.e. log transformation and various scaling) will significantly change absolute values, FC calculation are using data before the column-wise normalization was applied (i.e. at the original scale). The significant features are those features whose FCs are beyond the given FC threshold (either up or down).
Sorry - I originally answered with the convention for gene expression and proteomics data, not for metabolomics data. See Jeff’s answer.
1 Like
Hi,
Thanks for the questions and the answers on this topic. I had an additional question. Is there a reason why in metaboanalyst the FC is calculated based on the data before column-wise normalization and in express analyst the log transformed data is used for FC calculations?
Kind regards,
Lonneke
In transcriptomics, many tools (such as limma / edgeR) have their built-in normalization procedures coupled with differential analysis. These tools directly output logFC.
Of course, you can do the way as in MetaboAnalyst, but the mean estimations are not as accurate as those tools.