Hello,
I am a novice with proteomics, but I talked with a colleague about my proteomics normalization and he wanted to make sure that ExpressAnalyst is completing the QC order of operations properly for proteomics, i.e. Log2FC first then Normalization. I also believe that imputation should occur after normalization, but could be wrong? It seems some people perform different orders and Im not sure which is most appropriate.
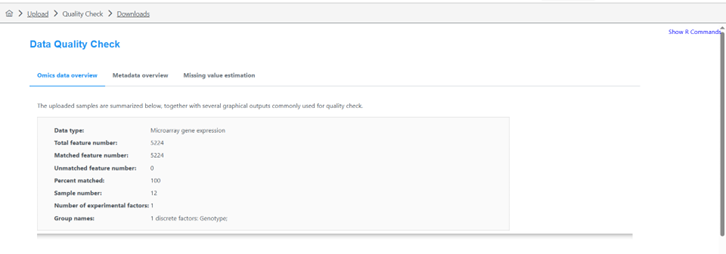
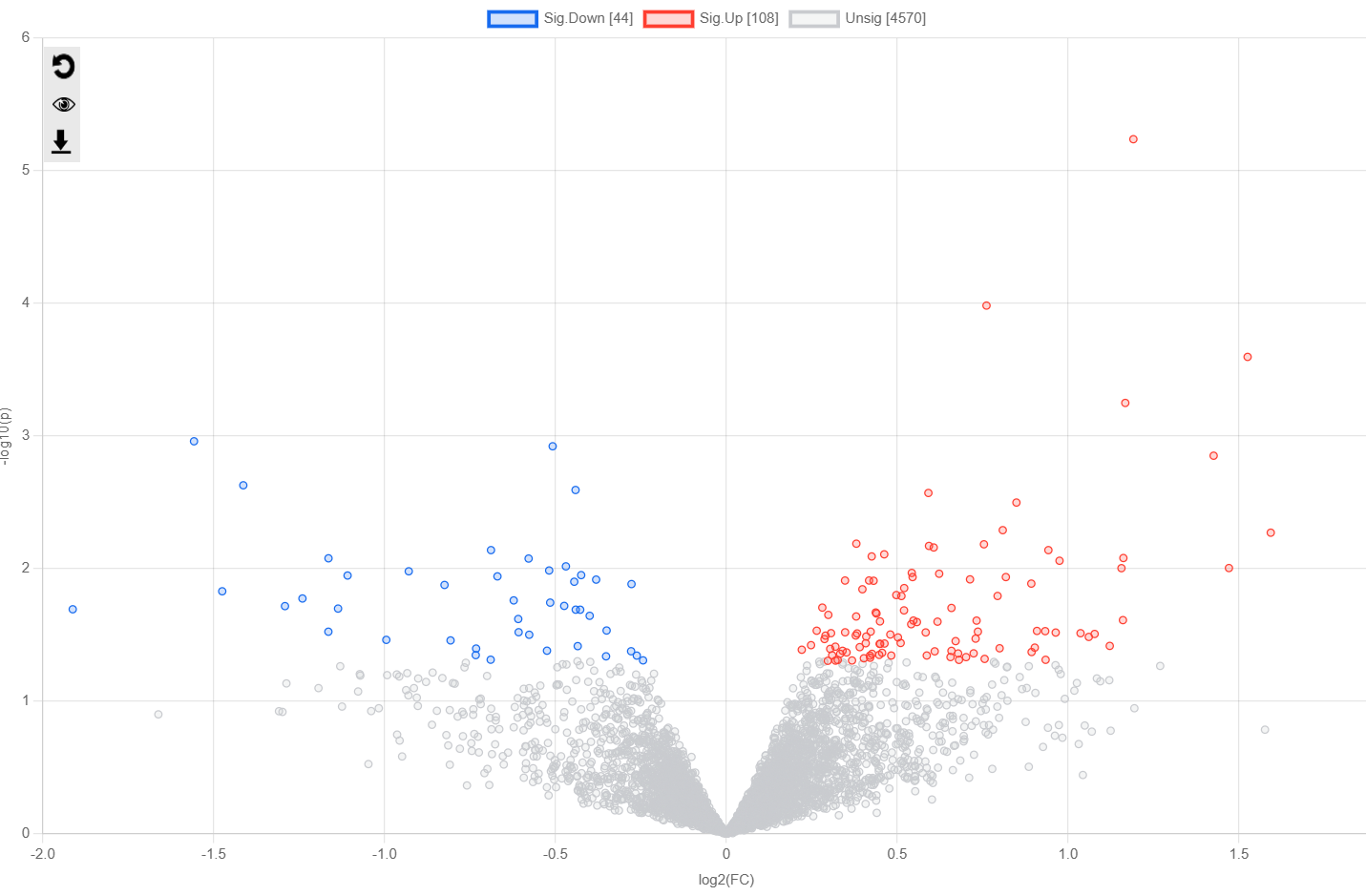
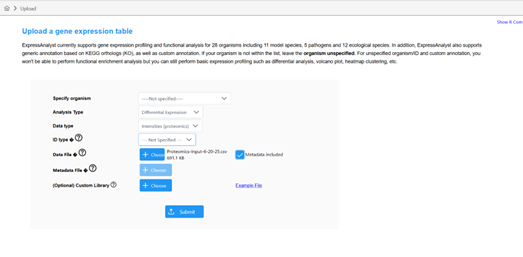
I saw in a previous post that this should be the case. HOWEVER, when uploading my dataset It is returning in the next ‘Data Quanity Check’ page as a MicroArray not a Proteomics dataset, even though I denoted ‘Proteomics’. I wanted to re-confirm that the software is returning the correct order of operations, as the R Command History does not provide much information on this?
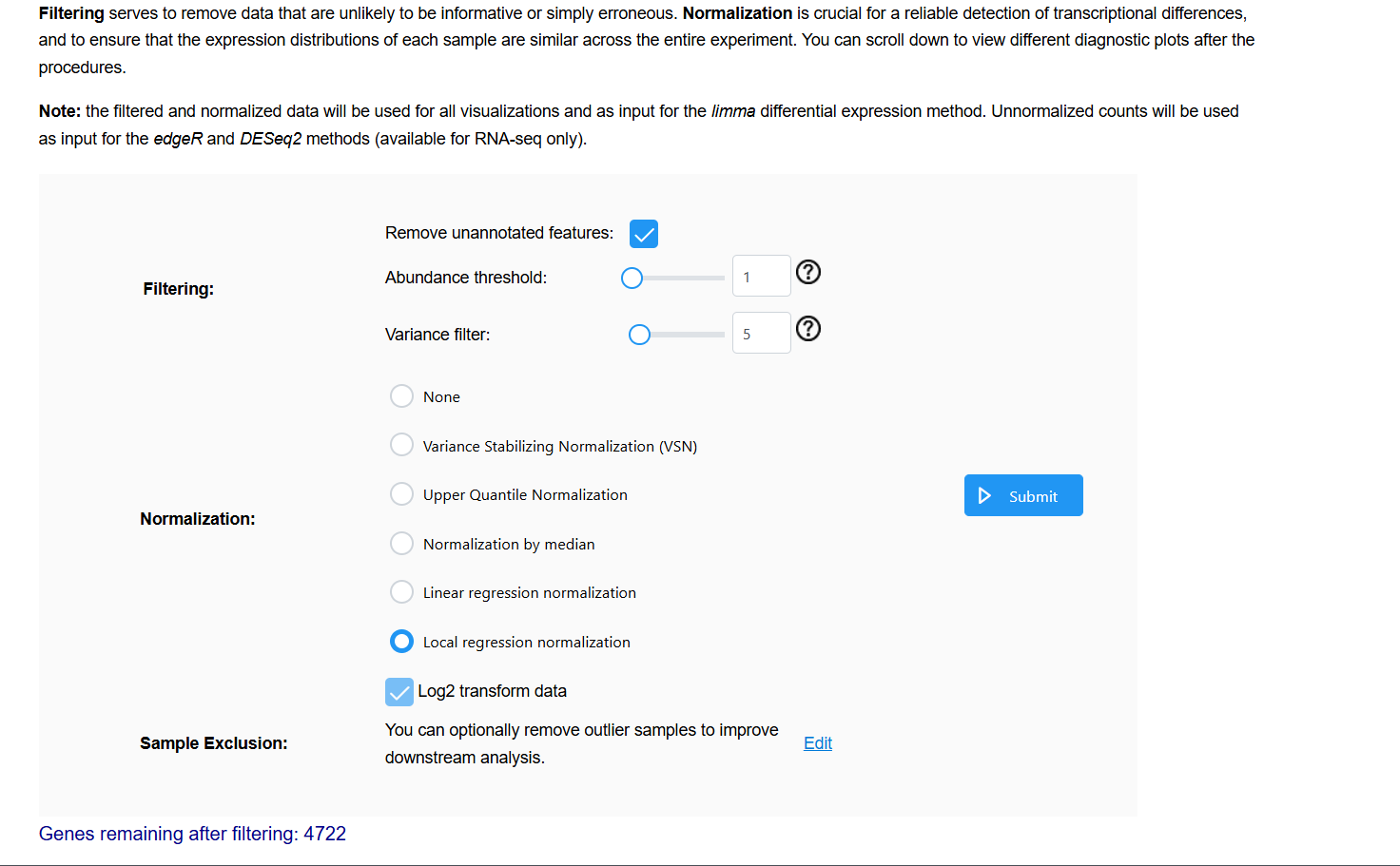
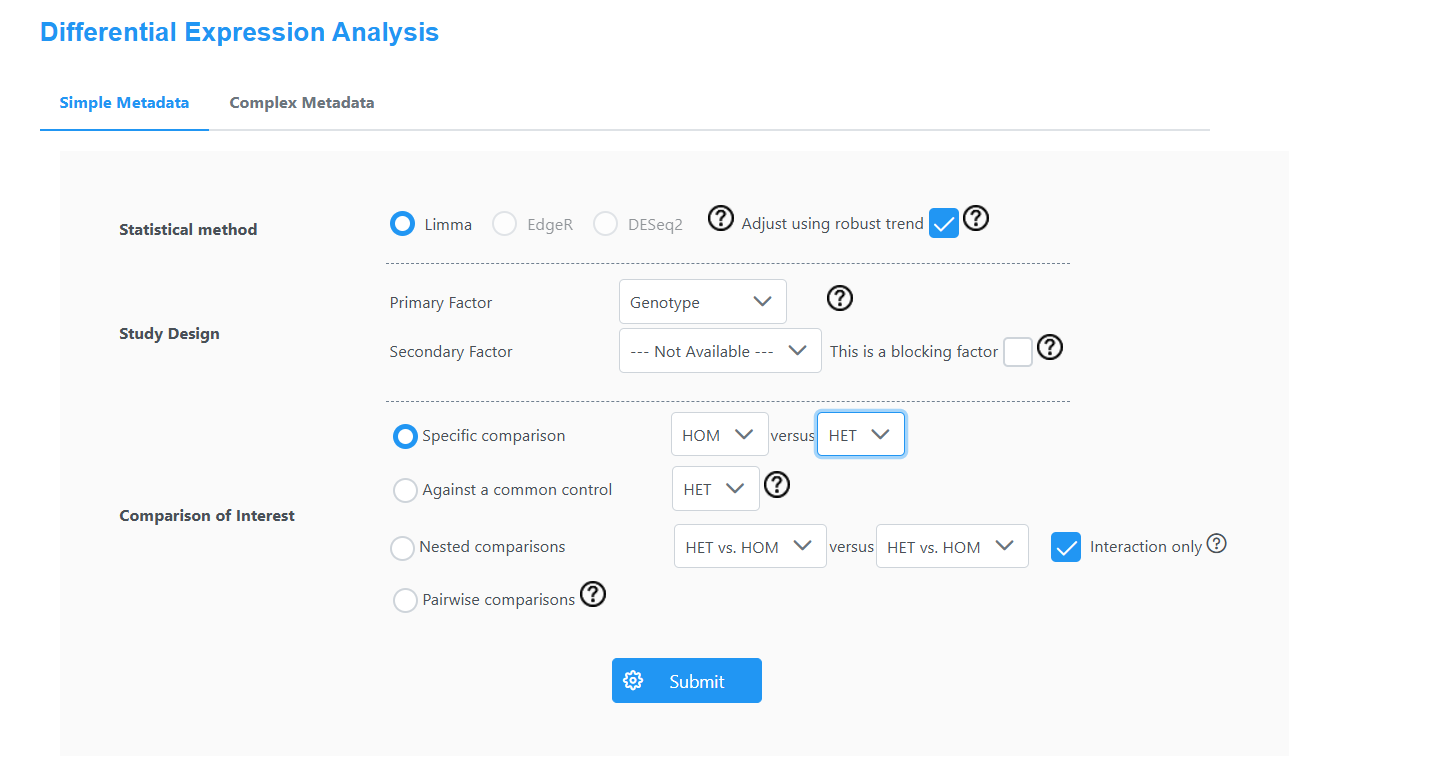
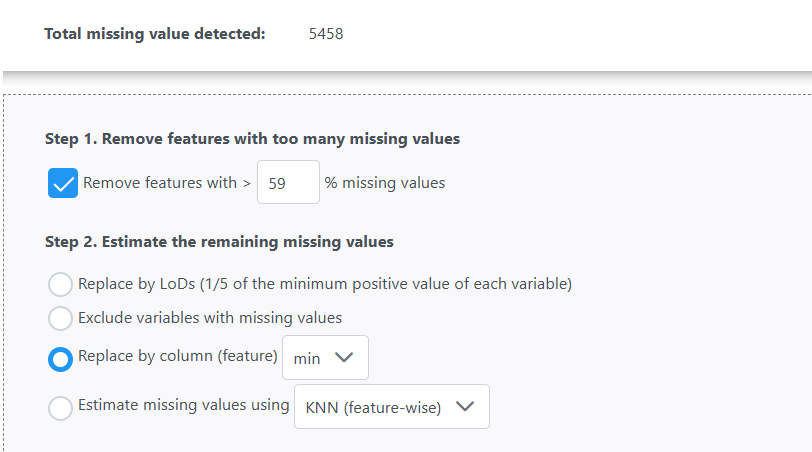
Please see the images below for my exact settings used.

For missing value estimation I used:
This is essentially removes anything with less than n=5 ( n=6 per group)
I am not very clear about your questions here. Some quick comments
-
You can analyze proteomics data using either ExpressAnalyst or MetaboAnalyst. However, there are some subtle differences: ExpressAnalyst follows Microarray/RNAseq practices. For instance, volcano plots and fold changes will be extracted from limma (microarray & proteomics), edgeR, DEseq outputs. MetaboAnalyst follows its own routine. It will directly use original values for FC analysis
-
R command order indicates logical steps, but the program has access to multiple copies of the data at different stages (original, filtered, normlized). It can use original data even after normalization step.
-
Should imputation be performed before or after normalization step? It depends. For instance, missing due to below the detection limits - they should be performed at original scale. In fact, many imputation methods will have its own built-in normlization procedures to ensure the performance (they will convert back to the original scale after imputation)