Dear MetaboAnalyst,
I have a question about log transformation in combination with the calculation of fold changes. I noticed that metaboanalyst uses the raw data for foldchange calculation and volcano plot generation even when you log transform the data beforehand. I read that this is because the fold changes change significantly after log transformation. However, I read on the internet that for proteomics and RNAseq data log transformation before fold change calculation is common. So, what is the difference and why did metaboanalyst choose not to do this for metabolomics data? Is there an inherent difference between metabolomics, proteomics, RNAseq data that is the reason for this?
Kind regards,
Lonneke
Volcano plot uses the values from Fold Change (FC) analysis, or call the same functions if it is not performed.
Note:
- FC reflects biological response (or effect size) and should use the values close to biological conditions. In your case, PQN (or any sample-wise or biological normalization) will have effect on FC, but not transformation nor scaling.
- Performing log transformation is common practice (this is used by MetaboAnalyst), as Log FC (A/B) = LogA - LogB
Hi,
I am sorry, but I do not understand your answer. In the picture below from the FC analysis from metaboanalyst it states that the original data is used for FC calculation , because column-wise normalization affects the absolute values significantly. So, do I understand correctly that also in the volcano plot the original data is used even if the user applied log2 transformation? In my hands, the log2 transformation also changes the log2 fold change a lot (Log2FC = mean(Log2 group A) - mean (Log2 group B)).
Maybe this post will help you. Note the first one is exactly your question. The second one clearly explains …
Thanks for the answer! But my question actually is, is there a reason why for metabolomics data in metaboanalyst the log2FC is calculated as Log2FC = log2(condition1) - log2(condition2) and for transcriptomics data/proteomics data in Express analyst Log2FC = mean(log2(condition1)) - mean(log2(condition2))? The change in calculation of log2FC changes the log2FC value and I am wondering why the calculation is different for the different types of data? Is there something inherently different between metabolomics data and transcriptomics/proteomics data (e.g. smaller changes in general)?
(Given that in both metaboanalyst and expressanalyst I opt for log2 transformation of my data prior to any analysis).
Can you clarify the source of this information? In transcriptomics, the logFC is usually obtained from limma / edgeR, we usually do not do this as in MetaboAnalyst.
Note you should not log2 transform your data before uploading your data to any of our tools.
Hi,
Okay, I did not know you should log2 transform before uploading. There is a normalization process in metaboanalyst. So that is not to be used?
I noticed that with my metabolomics data, the volcano plot stayed the same when I used my untransformed data versus the log2 transformation using the normalization tool in metaboanalyst. Then I read at the FC page, that for FC calculation the data before column wise normalization (in my case Log2 transformation) is used because the column wise normalization significantly changes the FC.
Then I started reading the internet, which mostly gives info for transcriptomics and proteomics data, that Log2FC = mean(log2(condition1)) - mean(log2(condition2)) is common. So, I wondered why this is common for these types of datasets, but metaboanalyst says the FC is altered significantly and that is why the original data is used. Why this difference?
Then I read the topic that I also commented on (Order of operation for fold change and p values - MetaboAnalyst - OmicsForum), from someone that had almost the same question as me. She put her data in metaboanalyst and there the FC calculation did not change when she log transformed in the normalization option of metaboanalyst. With express analyst, log transformation did change the FC, as she expected.
My question is: why this difference between metaboanalyst and expressanalyst? Is there a reason why in metaboanlayst it is specifically mentioned that FCs are calculated on non-columnwise normalized data, while in expressanalyst this does happen.
I hope this is clear.
Best,
Lonneke
Fold change is used to reflect biological response - the consideration here is what are true biological measures.
-
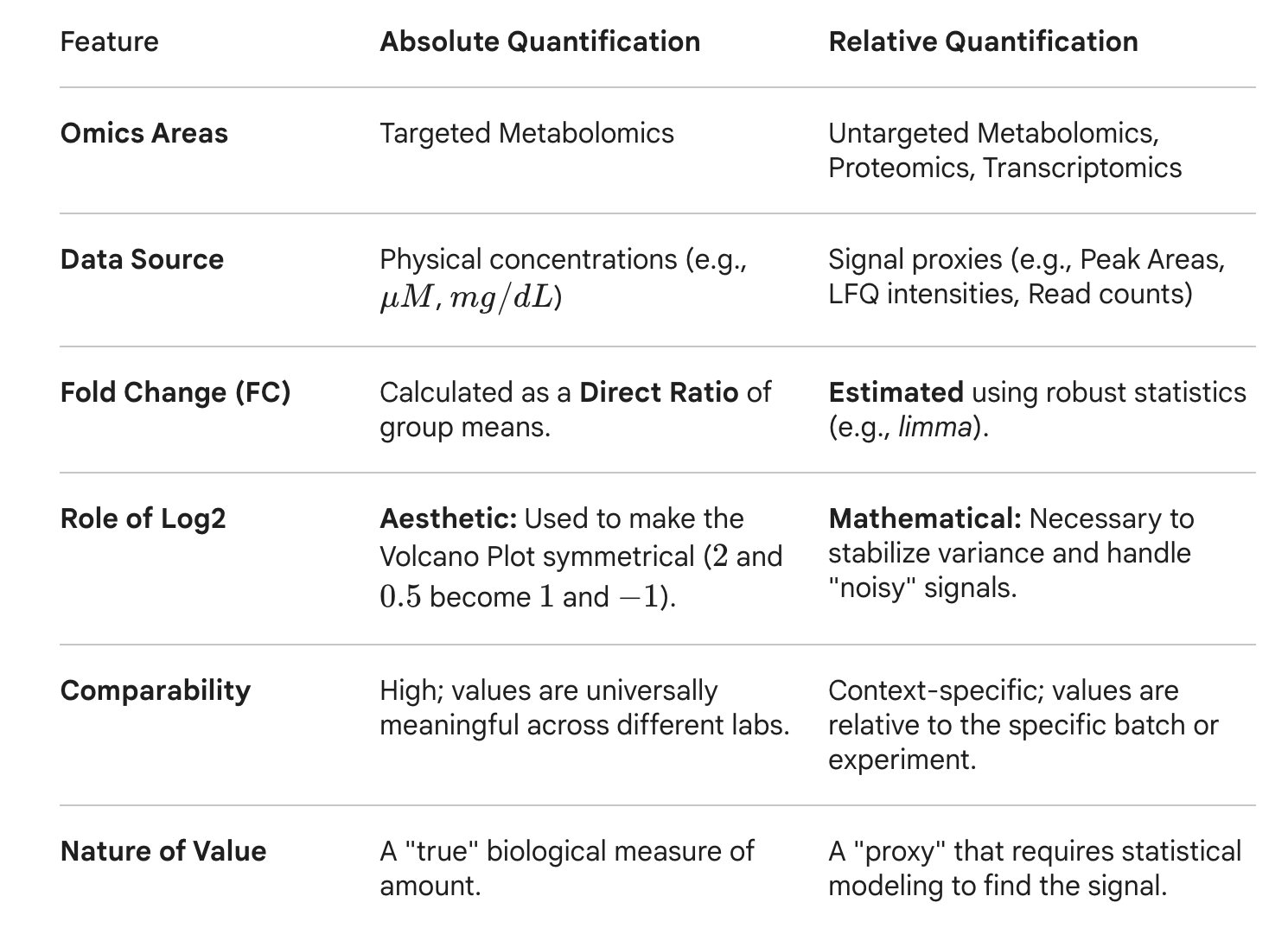
MetaboAnalyst was initially developed for targeted metabolomics data (i.e. absolute metabolite concentrations). The values are clinically meaningful and are universially comparable. For instance, we know the normal range of metabolite concentrations in human blood. The fold change is simply the ratios of two group means based on their concentration. Applying log2 is mainly aesthetic (i.e. 2 fold increase and 2 fold decrease are symmtrical ) so the volcano plot looks like volcano shape …
-
For other areas (including untargeted metabolomics) - the abundance values are proxies (i.e. peak areas, counts, intensities, cpms) and instrument- and data- specific. When sample size is small, the mean estimation are also very variable and unstable. Log transformation isn’t just for the plot – it’s for mean-variance stabilization. For instance, limma is often used to estimate fold change which is superior because it uses “empirical Bayes” to borrow information across all features.
I hope you see the differences. Below is a summary table (done by Gemini). Giving the questions, we intend to provide both options for MetaboAnalyst FC in the next update.
Thanks for answering! This answers my question. And sorry for misreading the ‘not’ in one of your answers.
Kind regards,
Lonneke