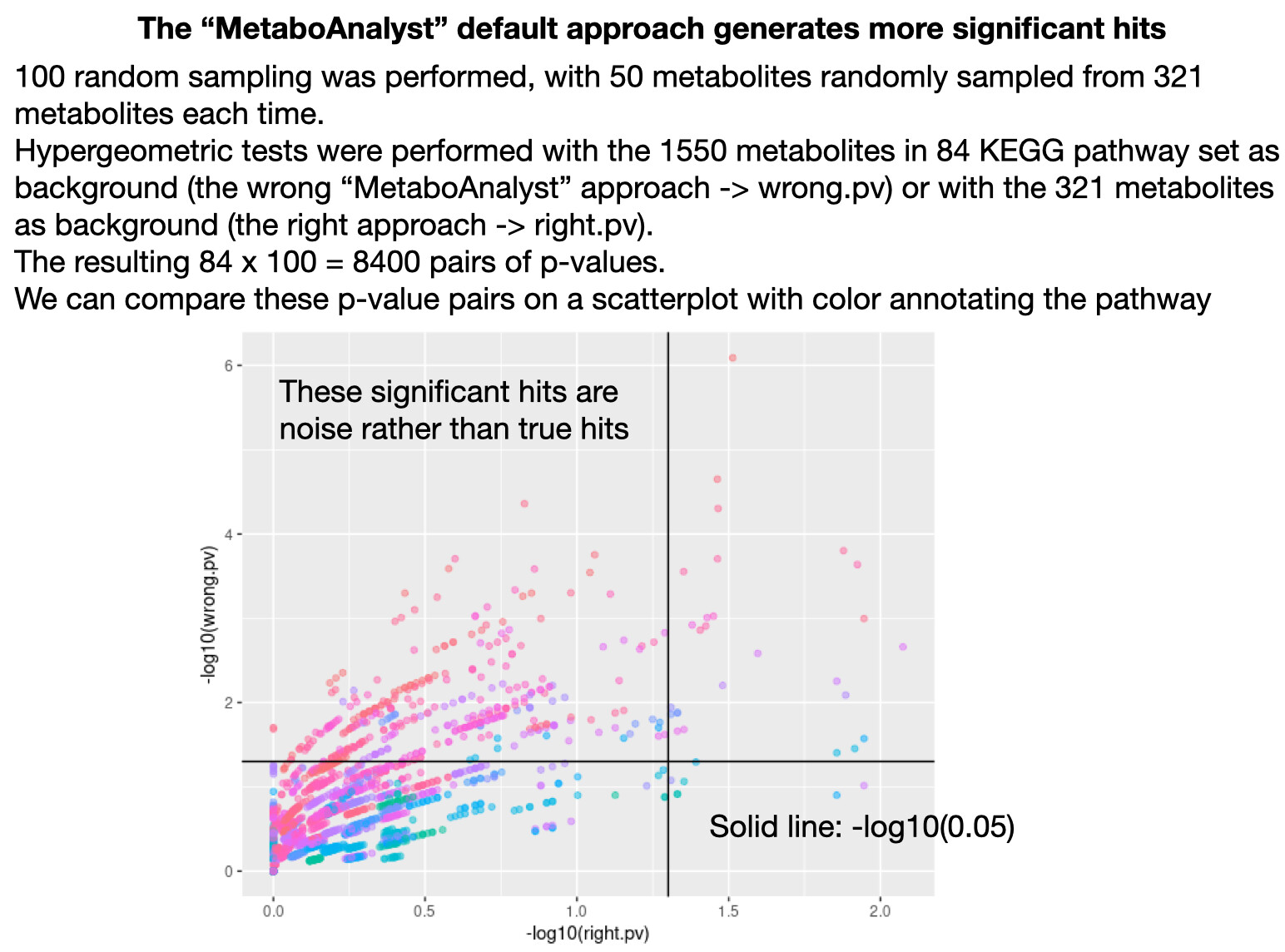
“Use all compounds in the selected pathway library” is provided as the default option for reference metabolome in the enrichment and pathway analysis module. However, unlike the other omics data, pathway coverage for metabolomics data are usually not even across different pathways. Metabolites that are lost during extraction procedures, too polar or non-polar for the LC, too labile for MS, etc won’t get detected. Using all compounds in the selected pathway library as the reference metabolome will wrongly pick up the pathways with high coverage as significant hits. The only correct option should be “Upload a reference metabolome based on your analytical platform”. However MetaboAnalyst has a bug that prevents this tool from running properly (see GitHub Issues 34,96 and 168). Please get these corrected soon. Thank you.
Thanks for the feedback! This is a valid concern and MetaboAnalyst has mentioned this potential bias from the day 1 (and in almost all of our tutorials on this topic).
For targeted metabolomics, MetaboAnalyst offers the option for “uploading a reference metabolome” in both Metabolite Set Enrichment Analysis module (it is working), and Metabolic Pathway Analysis module (will be fixed this week). For now, you can try the MSEA module which should give the same results (except the pathway visualization)
Thank you for the reply! I think the default option that generates biased results should be removed, since people (like me) may not be reading the tutorials and many will prefer methods that gives them statistically significant results rather than unbiased results. Attaching a test FYI.
MetaboAnalyst web platform is designed to allow users to explore their data interactively. For targeted metabolomics (i.e, ~40 compounds measured, 5 significant), there will be very few left after prefiltering. When cautions are taken, many researchers will ignore those “bias” or “obvious” pathway hits, or use them for quality assurance - they will continue down the list to find more meaningful hits.
When more compounds are measured (~100s), we can do better in algorithm. We are developing an unbiased approach, which does not require uploading a separate reference metabolome. Users will need to upload the full ranked list (or complete concentration table), and MetaboAnalyst will leverage this as background reference and further use permutation to compute empirical p values. This is similar to the mummichog approach currently employed for untargeted metabolomics. We hope to release this feature by the end of the year, if not earlier
Thank you. If the misleading option has to stay in the module, I think it should not be the default and should have warnings on the webpage (instead of the tutorial) to remind users to take cautions.
Looking forward to the new approach!