I understand why I should provide a reference metabolome when doing over-representation analysis. But why is there an option to provide one when doing quenatitative enrichment analysis? The data I upload contains intensities of all annotated metabolites, so the background should be defined with my dataset. Is it really necessary to provide a reference metabolome when doing QEA? Previous answers to similar questions are contradictory: Reference Metabolome for MetaboAnalystR and Why should I use a reference metabolome?.
Thank you for the question. I can see your perspective.
In MetaboAnalyst, when you upload your reference metabolome, it will be used to filter out the metabolite set library. Otherwise, the reference background is assumed to be all metabolites from the metabolite set library.
Since QEA is based on globaltest which is a self-contained method, the effect will be negligible, if any. Please feel free to share your experience / opinion
Thank you very much for your reply! Just to make sure I understand the filter aspect correctly: If a metabolite is present in the concentration data I supply but not in the reference metabolome, would it be considered for QEA or would it be filtered out? I use KEGG identifiers for concentration data but have to supply HMDB names for my reference metabolome which sometimes leads to problems due to imperfect mappings.
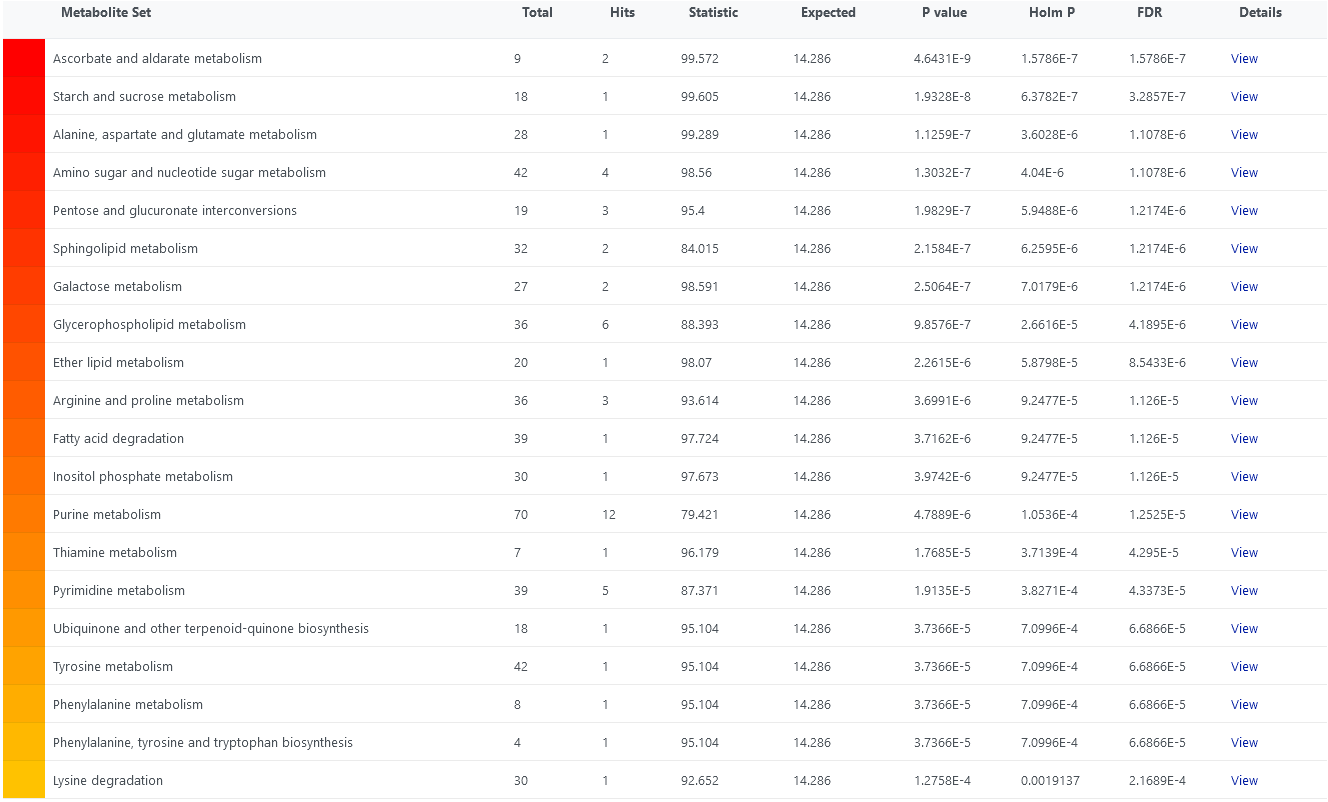
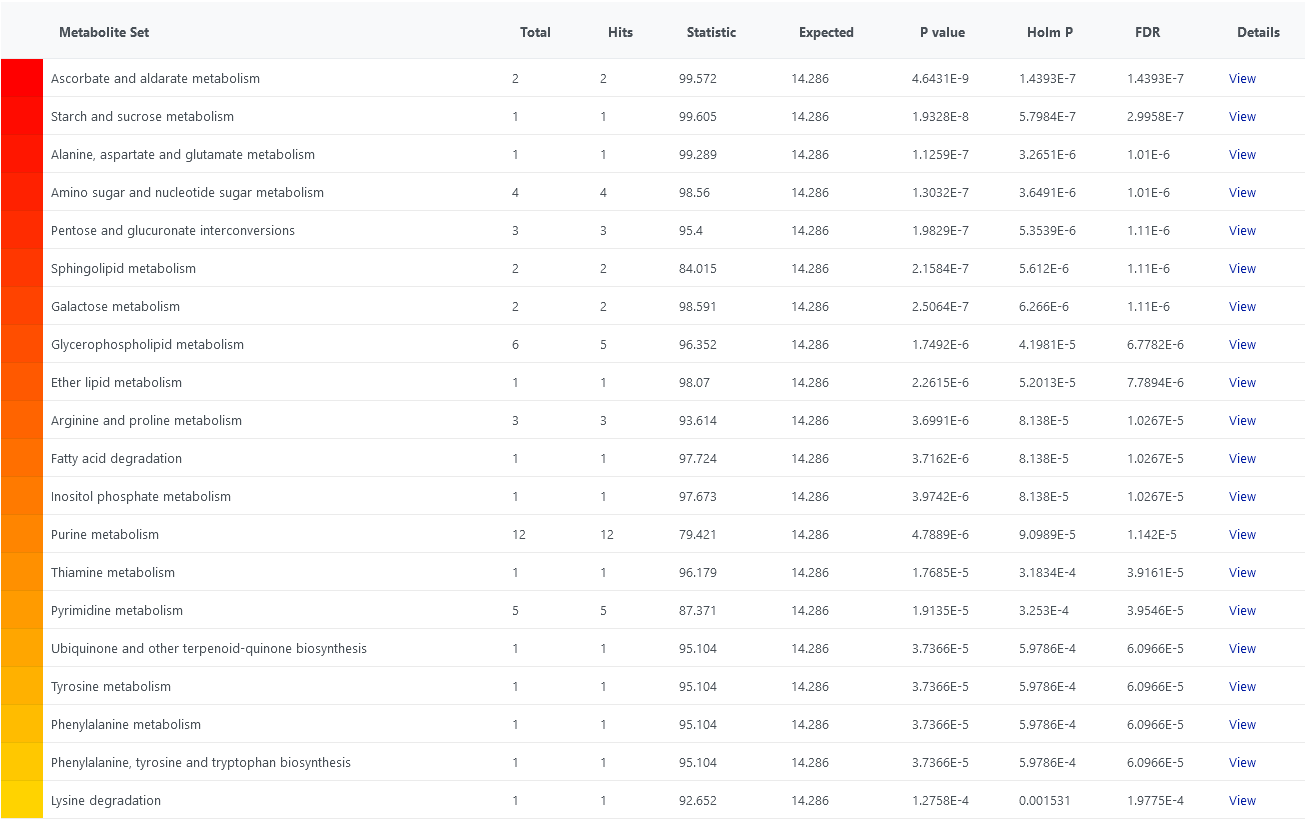
Also, I tested it with a data set I have prepared. I have attached the top 20 pathways. In the first image, I did not supply a reference metabolome and in the second one I did. For most pathways raw p-values are the same, except for ‘Glycerophospholipid metabolism’. Maybe that is due to the mapping problems I mentioned above? Also, corrected p-values are slightly different, probably because the list of pathways I got when I supplied a reference metabolome contains 3 pathways less than that where I did not supply one.
The behaviour is a bit strange in my opinion. Do you think it would help if there was an option to supply the reference metabolome in the same format (e.g., KEGG identifiers) as the concentration data/significant metabolites (in the case of ora)?
For now, I am more interested in raw p-values so I think I will stick to not supplying a reference metabolome. Anyways thank you very much and I hope I could help!