Omics data is designed to provide global overview (somehow, at the sacrafice of accuracy of indiivdual features). The proper approaches should be multivariate methods such as PCA, heatmaps, PLS-DA etc.
Your questions are mainly for classical univariate statistical methods which are designed to work with a few features but a lot of samples/replicates. These methods are in general not very suitable for omics data and their assumptions cannot be properly addressed/validated using omics data. However, univariate statistical methods are usually robust and can provide useful information for quick assessement of data quality and main pattern, as well as for feature selection.
For your particular questions
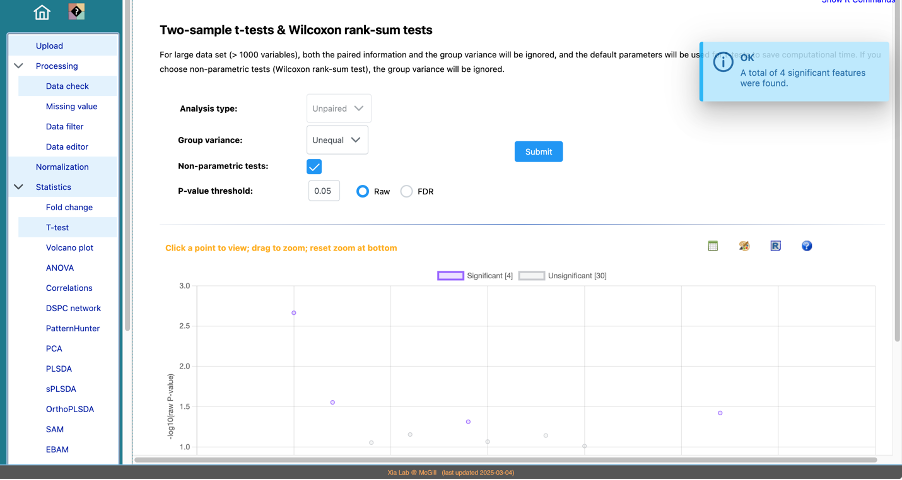
- The formula for group variance in t-tests are well defined - it literally refers to the variance of the group (not group size).
- In general omics data should be processed consistently (for instance, log transformation, auto-scaling applied to all features), not feature specific normalization after testing their individual normality
- Both raw (orignal p-values) and FDR (false discovery rate)-adjusted p-values are offered here as they are useful in different scenarios. If you use p-values as the key evidence for decision making, FDR (or the more stringent Bonferroni corrected p-values) is recommended
For univariate methods, linear modeling (limma) is designed for omics data than classifical t-tests / ANOVA. It is available in “Statistical Analysis [metadata table]” module. For more details, please refer to our nature protocol