PeaksToPathways_highvslowrisk_tscore_FINAL_clean.csv (730.2 KB)
PeaksToPathways_highvslowrisk_tscore.txt (730.2 KB)
Hello, My files for Pathway analysis are uploaded with this question. Can you please review and tell me the error? I think these are clean and align with the IBD2 example set.
Sorry for asking this elementary question.
thank you
Vidhu
Yes, I can see they are both OK, and also worked as expected (see below). Can you clarify what issue you experienced when you upload?
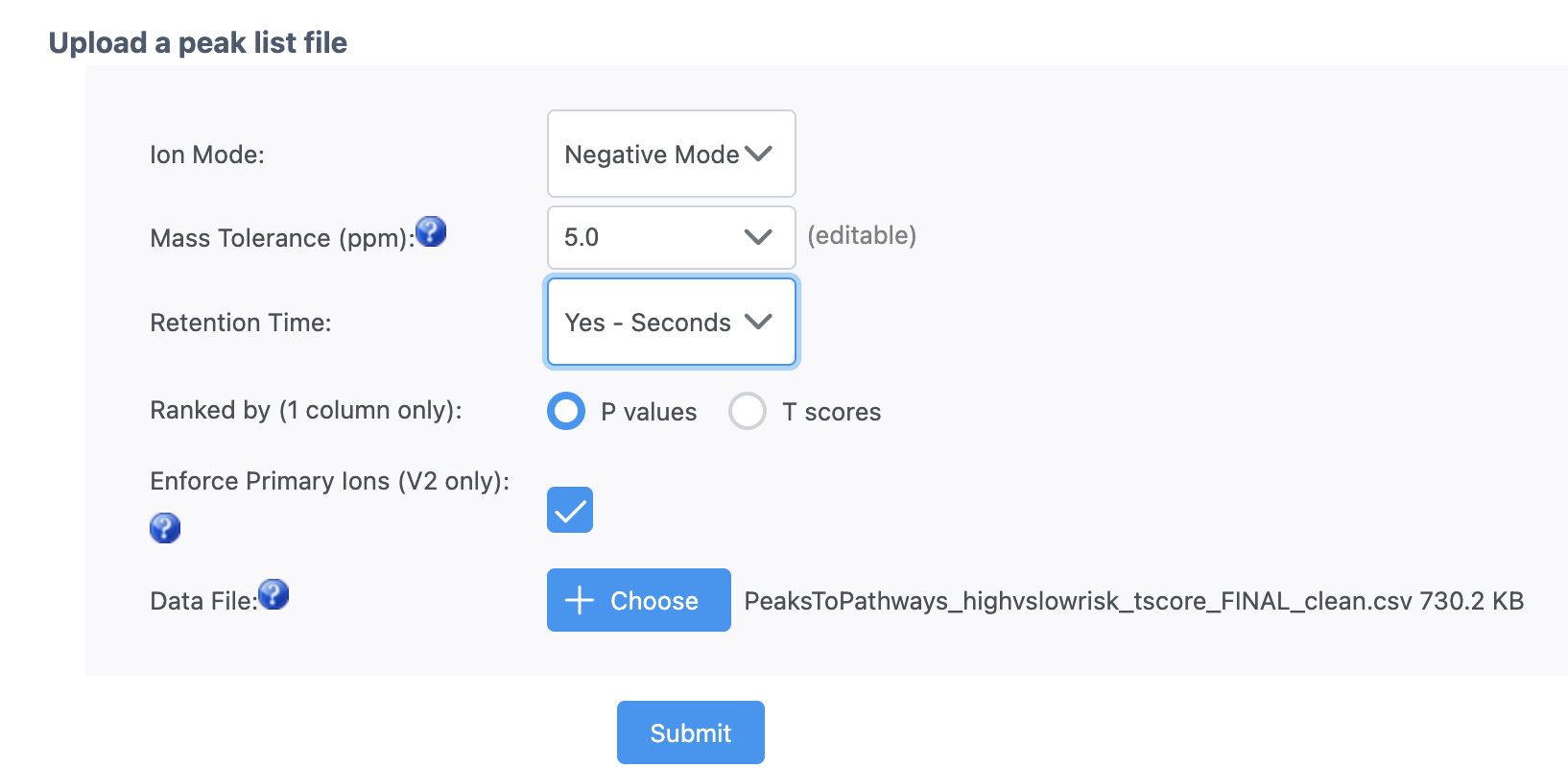
Thanks for the reply. Can you please try the mixed mode to see if it works for you? I have HILIC positive and C18 negative data in the sheet. Is it inappropriate to use mixed mode?
Mixed mode refers to positive & negative modes for the same column type. If you mix different columns, their retention times will not be comparable
Thank you for the response. Does this mean that I should upload HILIC positive and C18 negative separately? Does this also apply to RNA seq and metabolomics joint pathway analysis?
You can certainly upload and analyze HILIC positive and C18 negative separately - which is the default. Note we discussed several data integration strategies for meta-analysis or systems biology in our Omics Data Science course
- (low-level) Feature level integration
=> require data generated from the same sample from the same instrument - (middle) Compound level integration
=> require data generated from same samples. You annotate the data to compounds, and then combine them. This applies to different LC-MS instruments - (high) Pathway level integration
=> require data generated from the same tissue / individual. This applies to RNAseq and metabolomics integration
Dear Jeff and team,
I am analyzing joint pathway analysis again. The project involves placental RNA seq and cord blood metabolite profiling in the same mother-baby dyads. My project numbers are 7788 and 7790. In both of these, it appears that the transcriptome data was not utilized at all. Is there an error in the data upload or the workflow? Can there be other reasons preventing this processing? Functional analyses for the metabolites only has worked fine. Thank you
Vidhu
Analysis_Report.pdf (171.0 KB)
Hello Jeff and team,
For join pathway analysis, I am uploading a peaks list with MZ and RT and RNA seq data with official gene symbol and FC values. Attached is the result of the analysis. The plot suggests to me that only metabolites are being used for this analysis, the RNA seq is not being registered, or not being used in the analysis. Are you able to tell if there are errors in the workflow
thank you
Vidhu
Based on the Table 2, it seems that both genes and compounds were used. The issue is that the P values are all 1 for all gene based pathways . This is quite unusual.
Since you are doing untargeted metabolomics and RNAseq, pay attention to this difference
- metabolomic peaks should be the complete peak (per mummichog)
- gene names should only be the significant genes (per ORA)