Hi, how can I do a paired analysis between samples of differential relative abundance of OTUs in the MDP module (MicrobiomeAnalyst)? I want to compare the relative abundance before and after treatment, so need to pair samples - I can’t find any option for this under “single factor comparison”, only unpaired options . Would appreciate any advise. thanks.
You need to use the multi-factor analysis for this (you have two variables/factors: treatment and subject). To model your Sample ID as a random effect, specify it under ‘Blocking Factor’ and to model as fixed effect specify it under ‘Covariates (control for)’, and put your Treatment variable as ‘Primary Metadata’. Here is shown for modeling ‘Subject’ as a random effect for the example dataset #1:
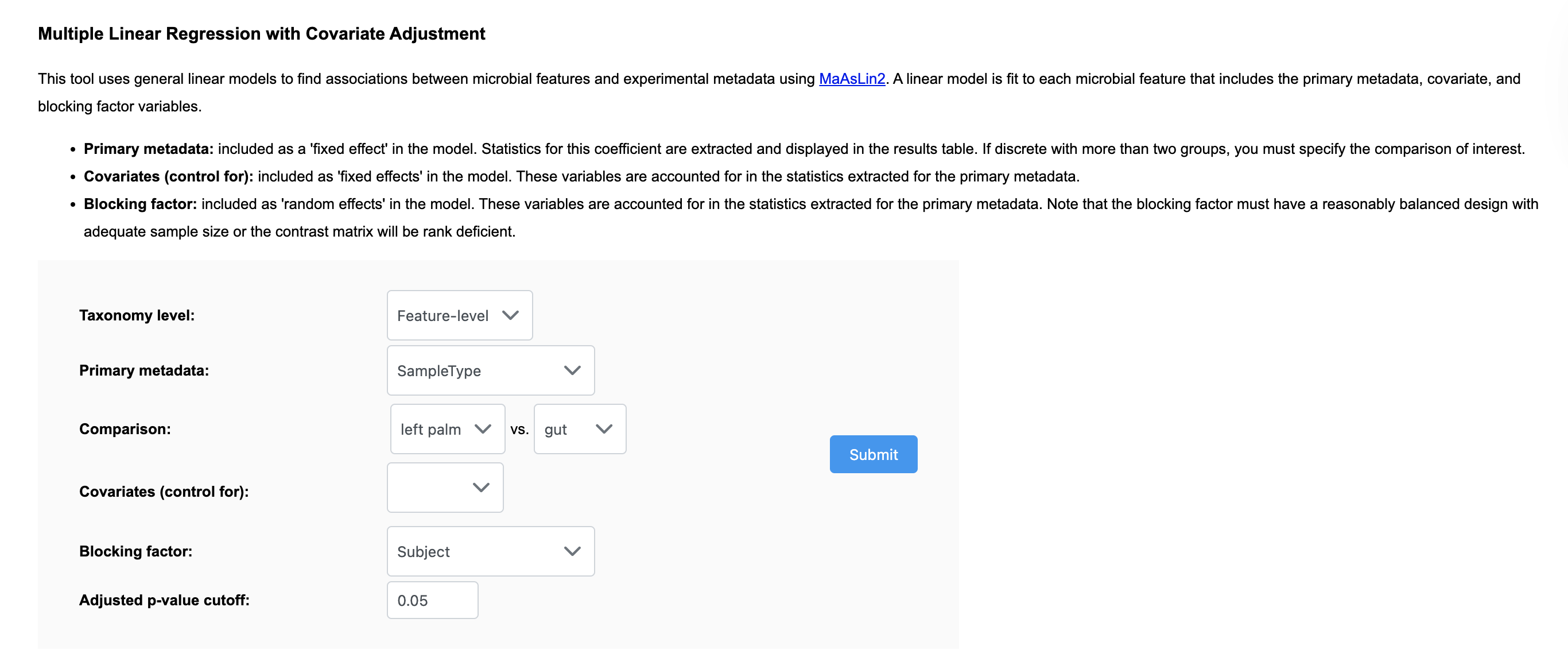
Specifying as a ‘Blocking Factor’ is more typical for paired analysis, however the R package we use has some more strict sample size/balanced dataset requirements for random effects modeling. If it’s not compatible with your dataset (you’ll get a message), try specifying as a ‘Covariate’ as this will also account for paired relationships.
when I add Subject to the meta data file (with the paired samples having the same subject in order to do the paired analysis as you wrote), the subject column doesn`t appear in the metadata file after upload - there are some subjects where there is no pair (just one sample) - is the whole column excluded because of that?
thanks
In order to control for subjects, the meta data file seems to have to include only paired samples - if there is 1 sample without a pair (like without a sample after treatment), then subject doesn’t appear as an option to chose as covariate. Is there a way to overcome this, since I want samples without a pair to be included in other analyses, besides the differential analysis?
Also, when using subject as covariant, is the comparison performed matched between each sample of a pair? meaning does it use the subject number to identify which samples to compare? The blocking option is not working as you expected.
Thanks
One practical solution is to create two different metadata files, using one for differential analysis (with the unpaired sample removed) and one for the other analyses (with all samples left in).
I would need more details to understand what you mean about “Blocking factor” (see our post guidelines)