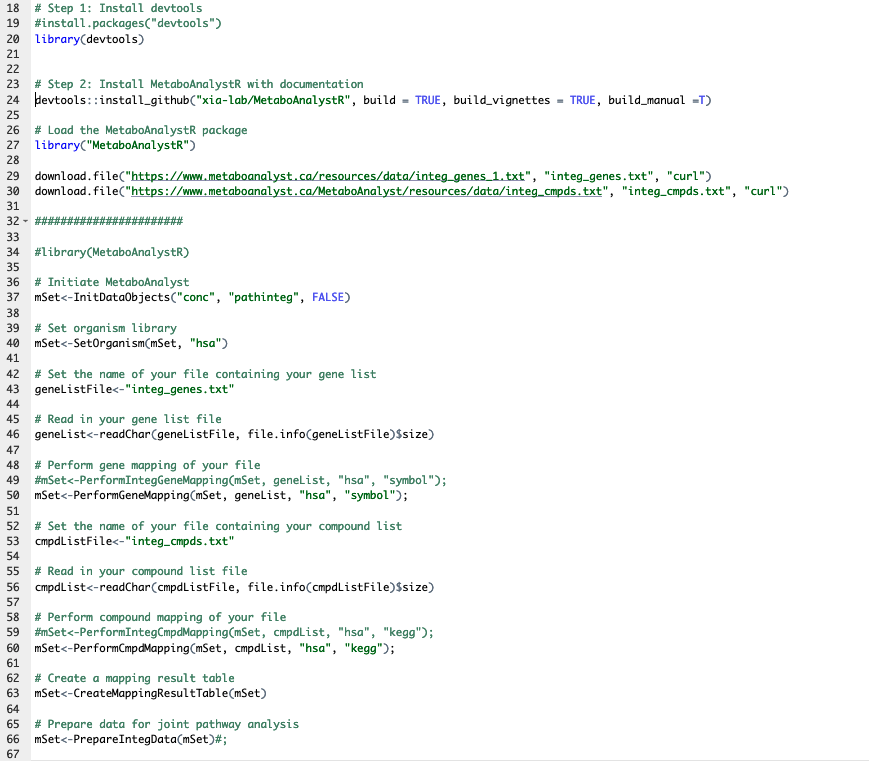
And I am having issues running the package. I also believe the PDF may be a bit outdated as there was a dead link and some misnamed functions, but after fixing the link and the misnamed functions the code still seems to run into errors. I am wondering if there is an updated R tutorial for Joint/Integrated Pathway Analysis or if there is a solution to the errors I am running into?
Here is the code I am currently running, that has been directly pulled from the PDF tutorial (with a few changes as I named above, line 29, 50, and 60):
Again, the files are directly downloaded from the Metaboanalyst and when the two files are run in the web-based tool there is no issue. Thanks in advance!
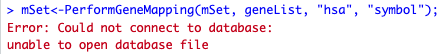
Thanks Qiang. I encountered the following error message when I run PerformGeneMapping() function:
mSet<-PerformGeneMapping(mSet, geneList, “hsa”, “symbol”);
Error: Could not connect to database:
unable to open database file
In addition: Warning message:
package ‘RSQLite’ was built under R version 4.1.3
I am using R version 4.1.1, do you think it’s the source of problem? In tutorial, it says R version 4.1 is compatible. But if not, I can update to 4.1.3 as shown in your sessionInfo() output. Thanks.
Hello Qiang, just upgrated to R 4.1.3, but still has the problem. (running a 64 bit R on Windows). It would great if you could help with this issue, thanks.
mSet<-PerformGeneMapping(mSet, geneList, “hsa”, “symbol”);
Error: Could not connect to database:
unable to open database file
Thanks for reporting this issue. I have fixed the database connection issue for MetaboAnalystR at Windows platform. Please update to the latest version and try again.
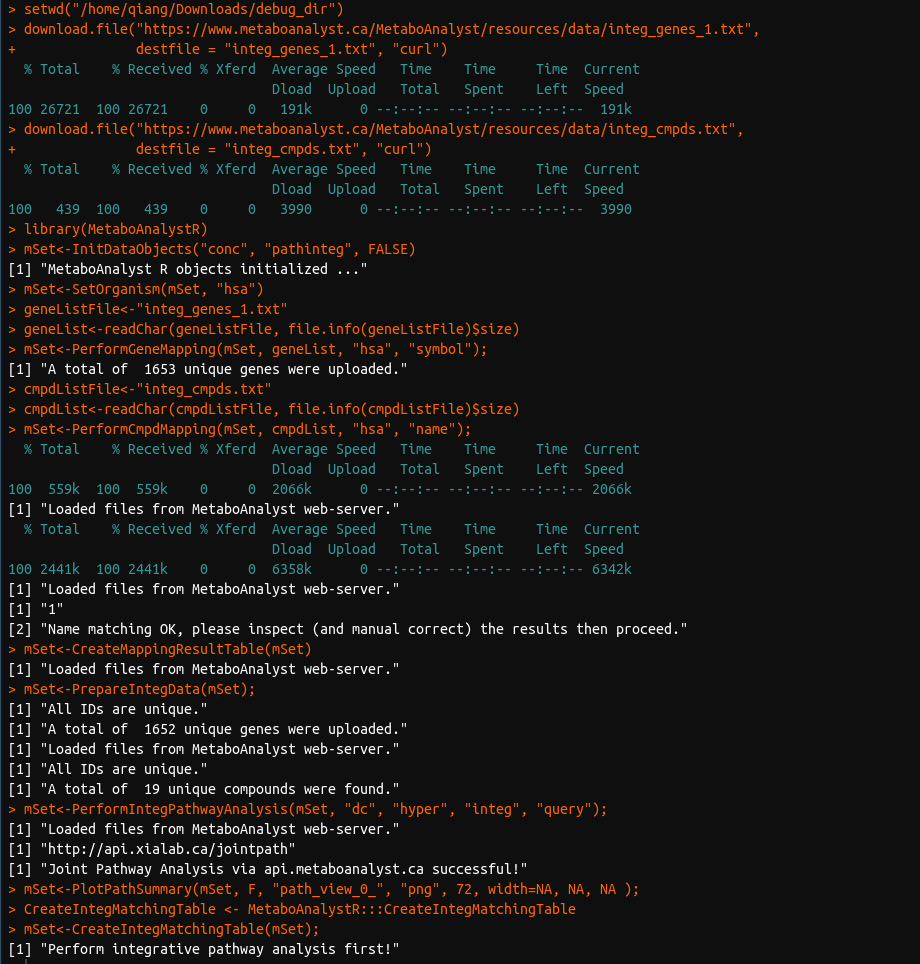
So I have updated to the newest version as you’ve advised Fred and it seems like the PerformGeneMapping() function now works, but the PerformCmpdMapping() still does not. Here is the code I am running:
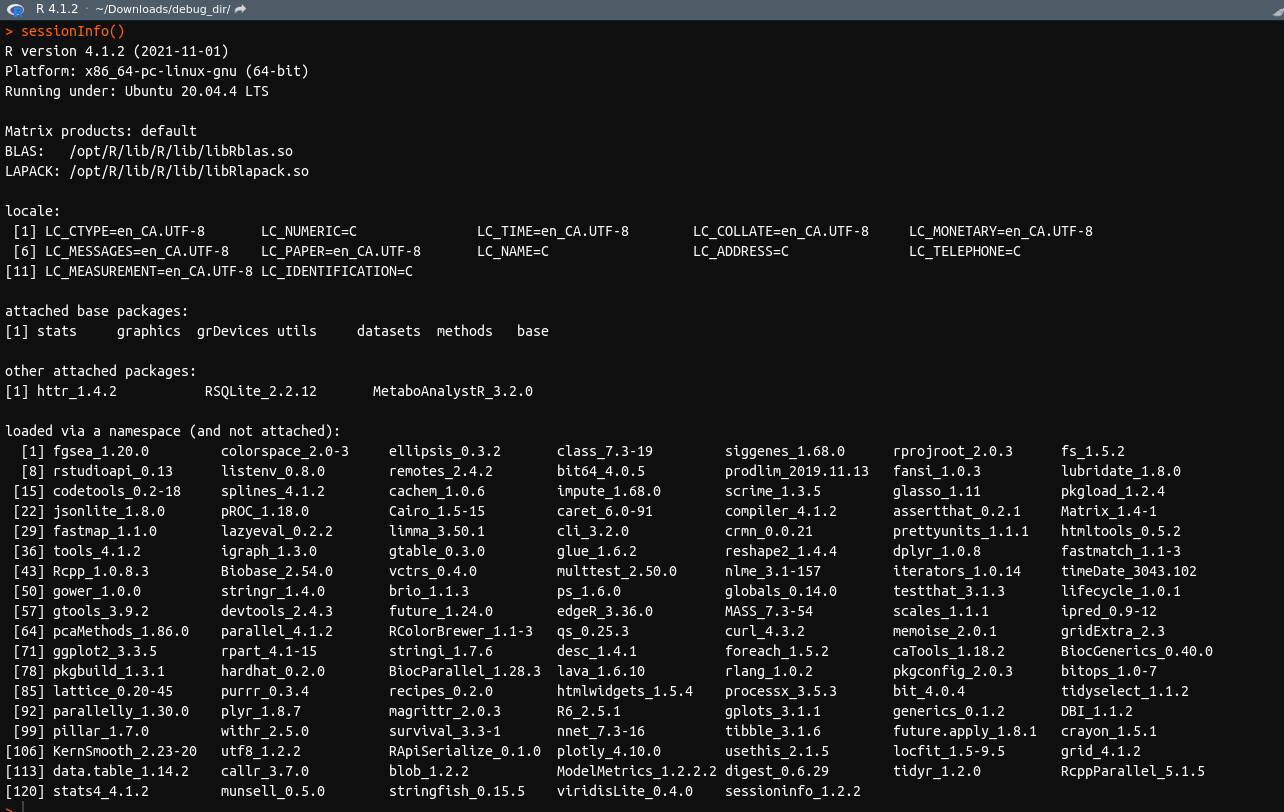
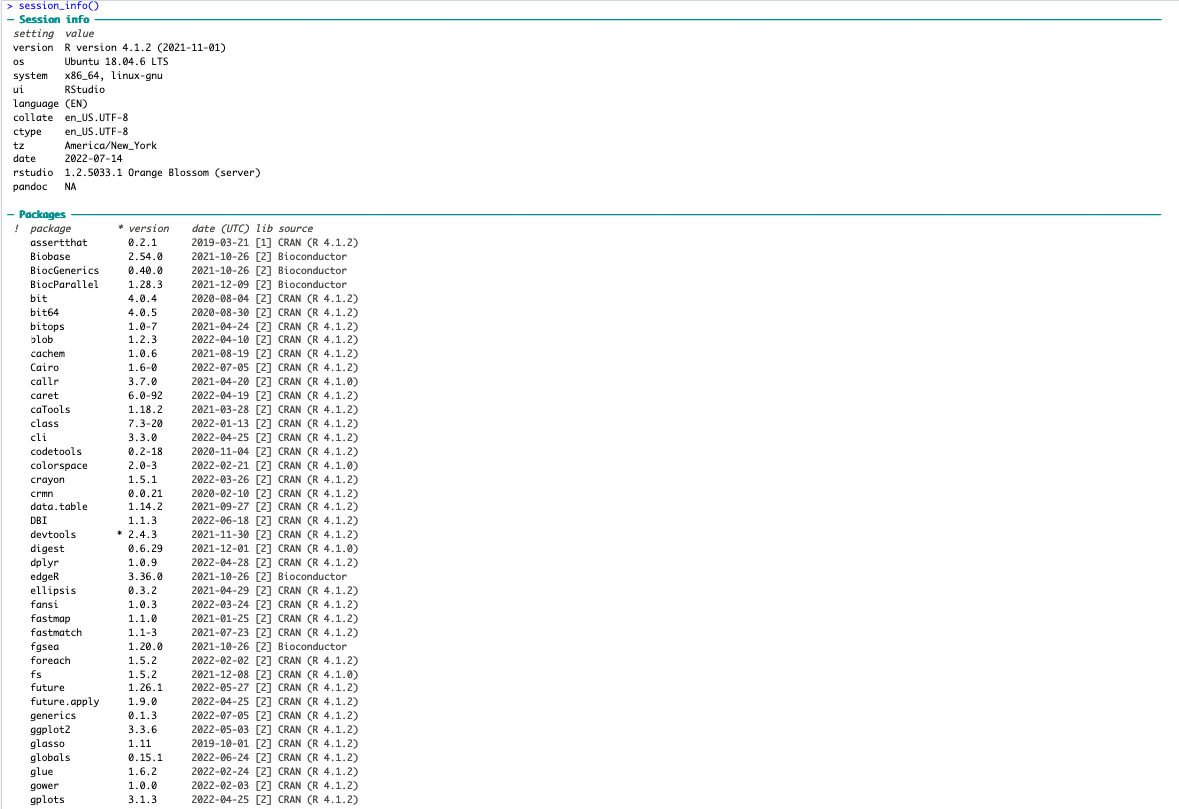
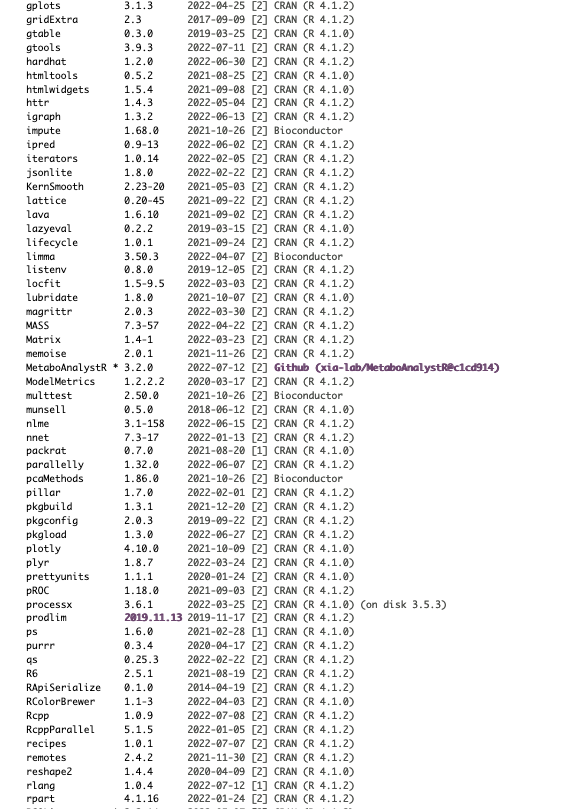
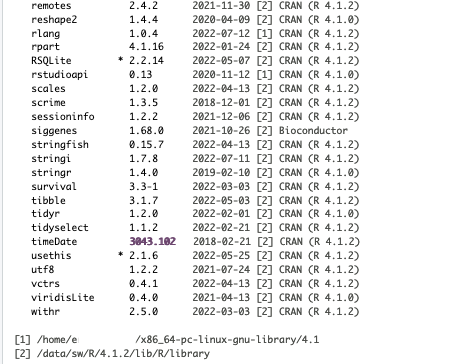
I am running all of this on R version 4.1.2 and pulling files directly from the Metaboanalyst website. Here is what my session info currently looks like:
I have recently tested this and it still seems like it is not working, even after updating to the latest version.
Again, PerformGeneMapping() seems to work, but PerformCmpdMapping() still does not work correctly as can be seen in my above post.
Wondering if you have any updates to this issue…
Thanks,
Erin
The issue is clear. For this command, you have to set the argument “idType” as “name” to run the whole srcipt, because the compound for the example data is compound name, rather than KEGG ID.