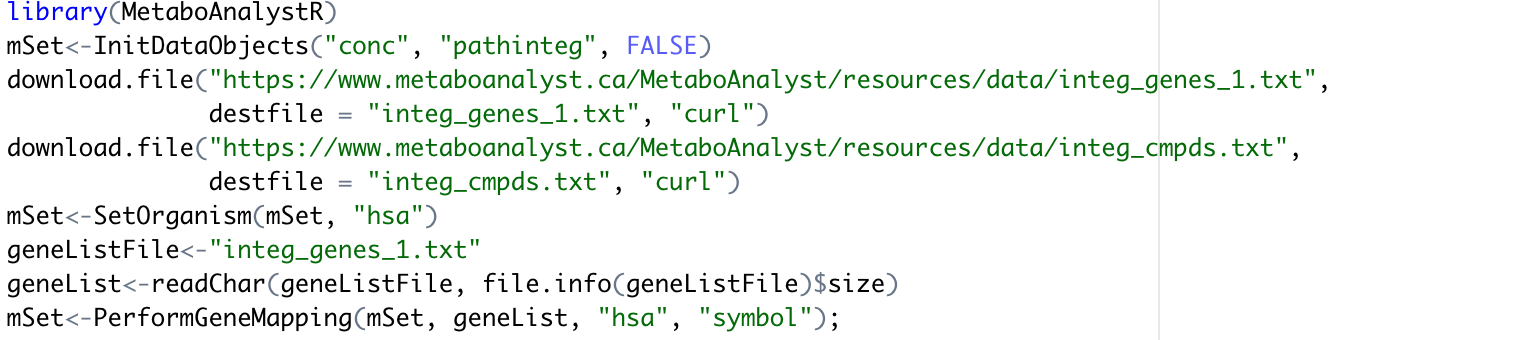
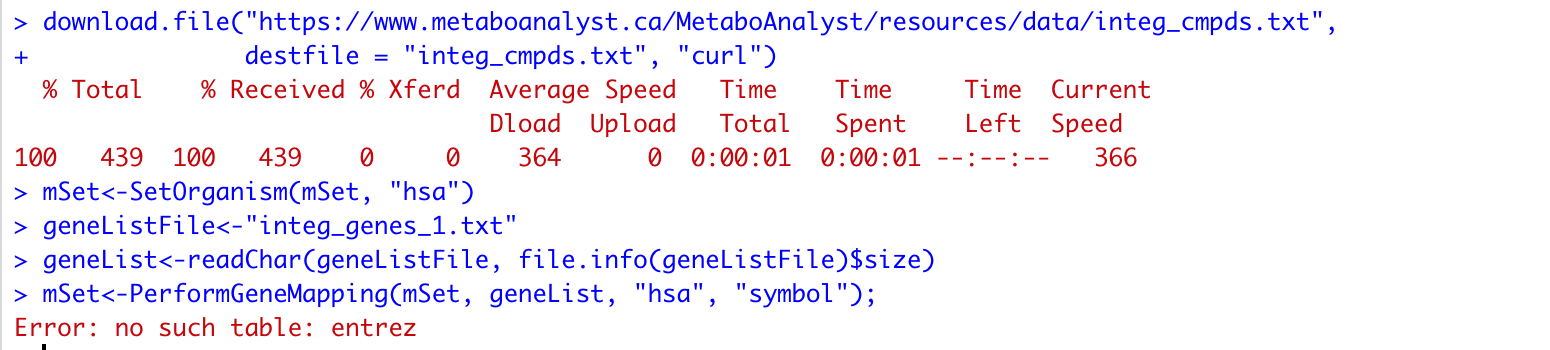
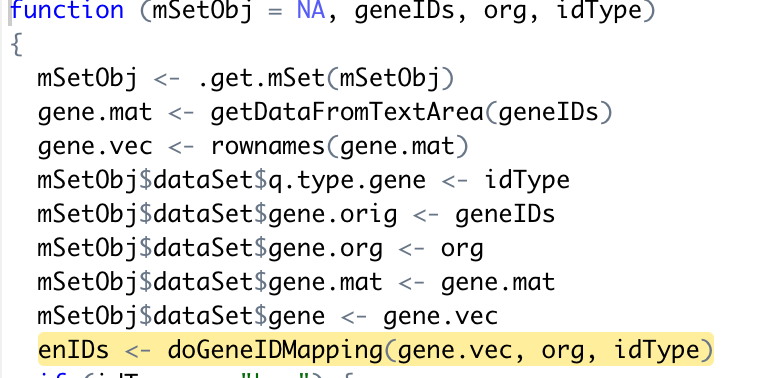
In the process of conducting joint-pathway analysis by MetaboAnalystR, there is an error raised when using the command mSet<-PerformGeneMapping(mSet, geneList, “hsa”, “symbol”); as ’ ‘Error: no such table: entrez’ '. Based my debug process, it seems to origin from the ‘doGeneIDMapping’ Function.
Here are my codes and session:
I tried for hours to solve the problem. These errors occur due to the lack of the hsa_genes.sqlite file in the working directory, which can be downloaded here:
doGeneIDMapping ← function(q.vec, org, type){
print(org)
sqlite.path ← paste0(url.pre, org, “_genes.sqlite”);
if(!file.exists(sqlite.path)){
#“https://www.xialab.ca/resources/sqlite/hsa_genes.sqlite”
sqlite_url ← paste0(“https://www.xialab.ca/resources/sqlite/”,
org, “_genes.sqlite”);
sqlite.path ← paste0(getwd(), “/”,org, “_genes.sqlite”)
download.file(sqlite_url,destfile = sqlite.path, method = “curl”)
}
Unfortunately, I am experiencing the same problem.
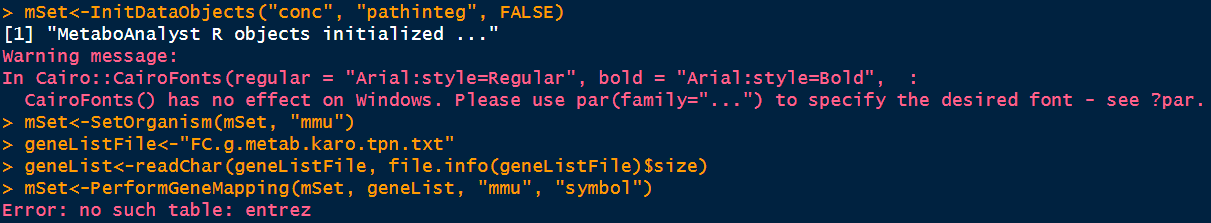
I want to run the joint pathway analysis for mice species, but I always get the Error: no such table: entrez. I tried the txt datafile example from the data input tab of the metaboanalyst webpage and a file containing only entrez IDs, but nothing seems to work. The weblinks in the comment also seem to be outdated?
How should I fix this issue?
Got the same problem. Compound mapping function works without error in R package. At the same time Web version reads Gene list just fine, but when I try to map genes with PerformGeneMapping() function in R it gives erroror (Error: no such table: entrez). Same thing with sample .txt files from https://www.metaboanalyst.ca/docs/Format.xhtml webpage.
This is my code: