When we perform the joint-analysis, if a pathway contains many more genes than metabolites, the significance of the genes will overwhelm the significance of the metabolites. In addition, the transcriptomics usually report far more differential expression genes than metabolomics, in this case, the integration result is always dominated by transcriptomics datasets.
The weight strategy for different ‘universes’ (“transcriptomics data universe” and “metabolomics data universe”) are performed with a weighted z-test. The weighted z-test proposed by Dmitri V. Zaykin. et. al is designed for the weighted integration of different datasets of very different sizes. A figure below is provided to illustrate the mechanism of this weighted integration of different Omics-data in MetaboAnalyst.
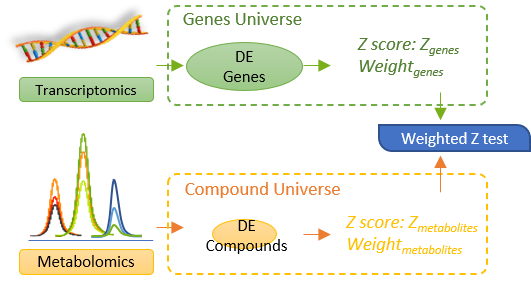
Specifically, we assign different weights based on the proportion of genes and metabolites in the specific ‘omics universes’ to balance the influence from the different sizes of the ‘omics inputs upon the integrated pathway results. The adjusted P value is estimated with a weighted Z-test below.
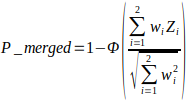
In the equation, wi is the weights of the P values of genes or compounds within individual omics “universe” or “pathway space”, respectively; Zi is the Z score of the corresponding P values of single omics data, usually, Zi = Φ−1(1 −Pi); Pi is the P values from the enrichment analysis above; Φ denote the standard normal cumulative distribution function.