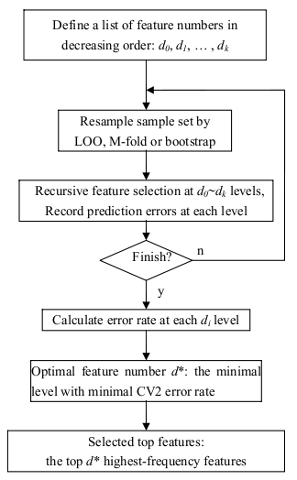
Recursive SVM (R-SVM) uses SVM for both classification and for selecting a subset of relevant genes according to their relative contribution in the classification. This process is done recursively so that a series of data subsets and classification models can be obtained in a recursive manner, at different levels of feature selection.
The performance of the classification can be evaluated either on an independent test data set or by cross validation on the same data set. R-SVM also includes an option for permutation experiments to assess the significance of the performance. Please note, only linear kernel was used for classification, since the information is usually far from sufficient for reliably estimating nonlinear relations for high-dimensional data with a small sample size. First-order approximation will reduce the risk of overfitting in case of limited data. CV2 refers to the cross validation embedded with the feature selection procedure as discussed above. The chart below summarize the R-SVM workflow. For more details, please read the paper by Zhang X, et al.