Hi all! First thing, big fan of Metaboanalyst! It has helped my research for many projects
I am trying to do some enrichment analysis using as a starting point metabolomics data generated by Metabolon from mouse plasma samples. So I have a list of metabolites (I have both HMDB and KEGG ids) after statistical analysis.
I have tried Enrichment and Pathway analysis and I get only a result when using universal libraries as a reference.
However, I would like to get a result using as a reference metabolome the list of metabolites identified in this dataset (regardless of significance).
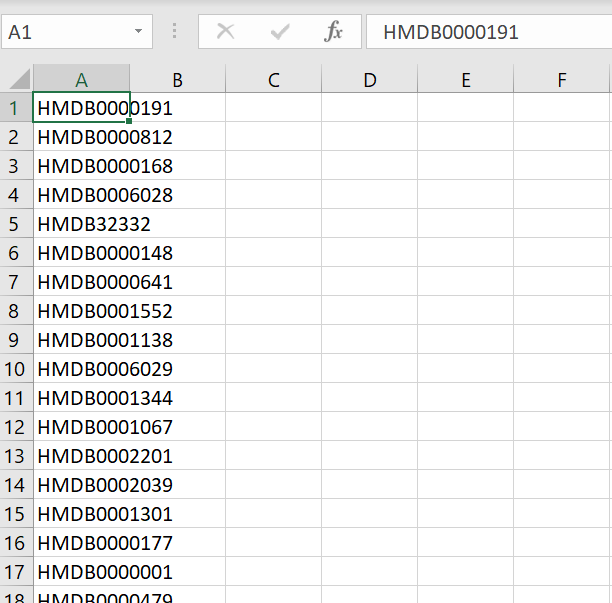
I created a single-column file with all the metabolites identified, like this:
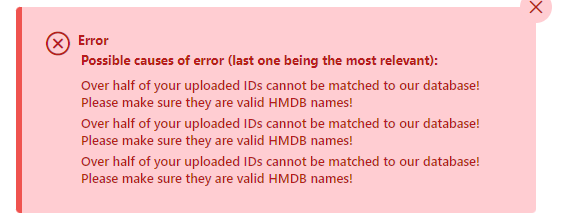
Thanks for the note. As indicated by the error message, there are a lot of compounds detected (i.e. your reference IDs), but their functional annotations (i.e. pathways in our libraries) are sparse. This is similar to limited gene ontology coverage in early days, will require a lot manual effort to address this issue.
We will soon add a new algorithm to take account of the reference metabolome in detecting enriched pathways. The approach should be more robust than the current one