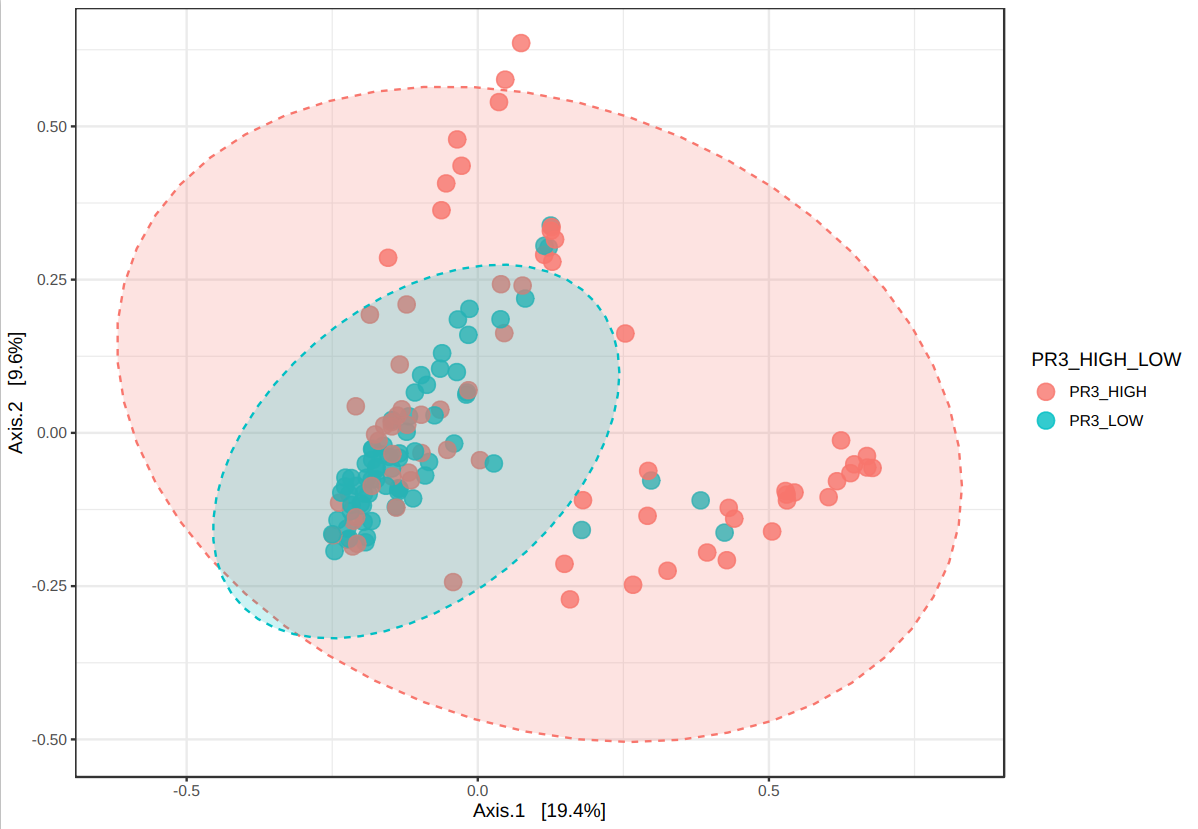
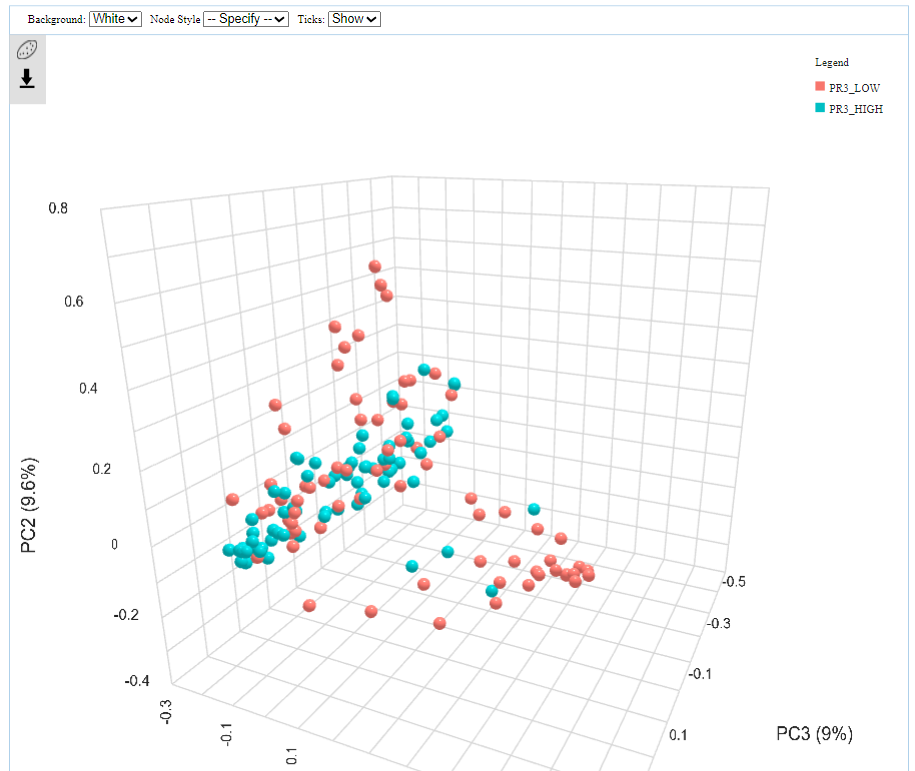
I have been performing beta diversity analysis in microbiomeanalyst for several biomarkers using PERMANOVA. I noticed that, in the 3D plot, the marker labels are switched (ie showing the opposite results from the 2D plot). I have pasted an example below as well as the R code. This is the same for 4 different markers I have tried. I wonder if there is an error in the code that is causing the labels to be switched between the two plots?
| 1. | mbSet<-Init.mbSetObj() |
|---|---|
| 2. | mbSet<-SetModuleType(mbSet, “mdp”) |
| 3. | mbSet<-ReadSampleTable(mbSet, “emma-meta.csv”); |
| 4. | mbSet<-Read16STaxaTable(mbSet, “LTBE.csv”); |
| 5. | mbSet<-Read16SAbundData(mbSet, “EmmaOTUfinal.csv”,“text”,“SILVA”,“T”,“false”); |
| 6. | mbSet<-SanityCheckData(mbSet, “text”); |
| 7. | mbSet<-SanityCheckSampleData(mbSet); |
| 8. | mbSet<-SetMetaAttributes(mbSet, “1”) |
| 9. | mbSet<-PlotLibSizeView(mbSet, “norm_libsizes_0”,“png”); |
| 10. | mbSet<-CreatePhyloseqObj(mbSet, “text”,“SILVA”,“F” , “false”) |
| 11. | mbSet<-ApplyAbundanceFilter(mbSet, “prevalence”, 0, 0.1); |
| 12. | mbSet<-ApplyVarianceFilter(mbSet, “iqr”, 0.0); |
| 13. | mbSet<-PerformNormalization(mbSet, “none”, “none”, “none”, “true”); |
| 14. | mbSet<-PerformBetaDiversity(mbSet, “beta_diver_0”,“PCoA”,“bray”,“expfac”,“PR3_HIGH_LOW”,“none”,“OTU”,“”,“Chao1”, “yes”, “adonis”, “png”, 72, “default”); |
| 15. | mbSet<-PCoA3D.Anal(mbSet, “PCoA”,“bray”,“OTU”,“expfac”,“PR3_HIGH_LOW”,“”,“Chao1”,“beta_diver3d_0.json”) |
| 16. | mbSet<-PerformCategoryComp(mbSet, “OTU”, “adonis”,“bray”,“PR3_HIGH_LOW”); |