Hello,
I have been working on running a few data sets of FASTQ files for species without annotated genomes through Seq2Fun using Docker Desktop. I have successfully run one species, and though I experienced one failure, it succeeded on the second attempt. However, when trying to run the second species, the run has failed 3 times. I am able to begin the run, it accepts the metadata and pathways, and gets through 8-12 of the 24 samples before stopping. It stops at a different point each time.
I am inputting the fastq.gz files, and running on fish orthologs (fishes_v2.0.fmi and fishes_annotation_v2.0). I am using the dockerxialab/expressanalyst_docker:latest image, created 3 months ago. The runs are set to use 12 threads, with all other settings left at default.
Is it a matter of just re-starting the run until it succeeds, or is there some other trouble-shooting I can try?
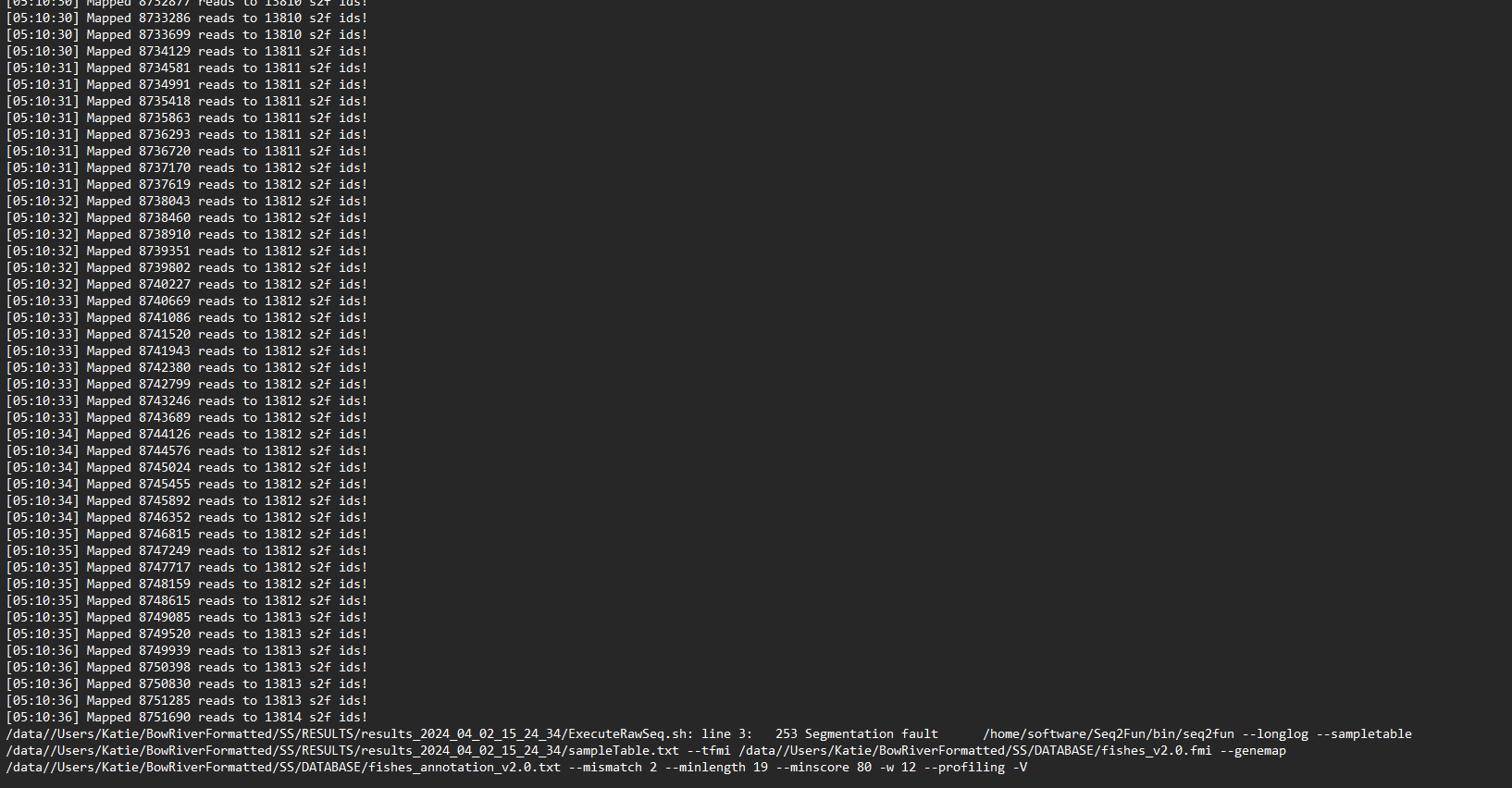
When checking the job_logs, the errors are as follows:
/data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_02_15_24_34/ExecuteRawSeq.sh: line 3: 253 Segmentation fault /home/software/Seq2Fun/bin/seq2fun --longlog --sampletable /data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_02_15_24_34/sampleTable.txt --tfmi
/data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_01_06_27_11/ExecuteRawSeq.sh: line 3: 275 Segmentation fault /home/software/Seq2Fun/bin/seq2fun --longlog --sampletable /data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_01_06_27_11/sampleTable.txt --tfmi
/data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_01_21_43_48/ExecuteRawSeq.sh: line 3: 262 Segmentation fault /home/software/Seq2Fun/bin/seq2fun --longlog --sampletable /data//Users/Katie/BowRiverFormatted/SS/RESULTS/results_2024_04_01_21_43_48/sampleTable.txt --tfmi
Cheers!
Katie
Files below: