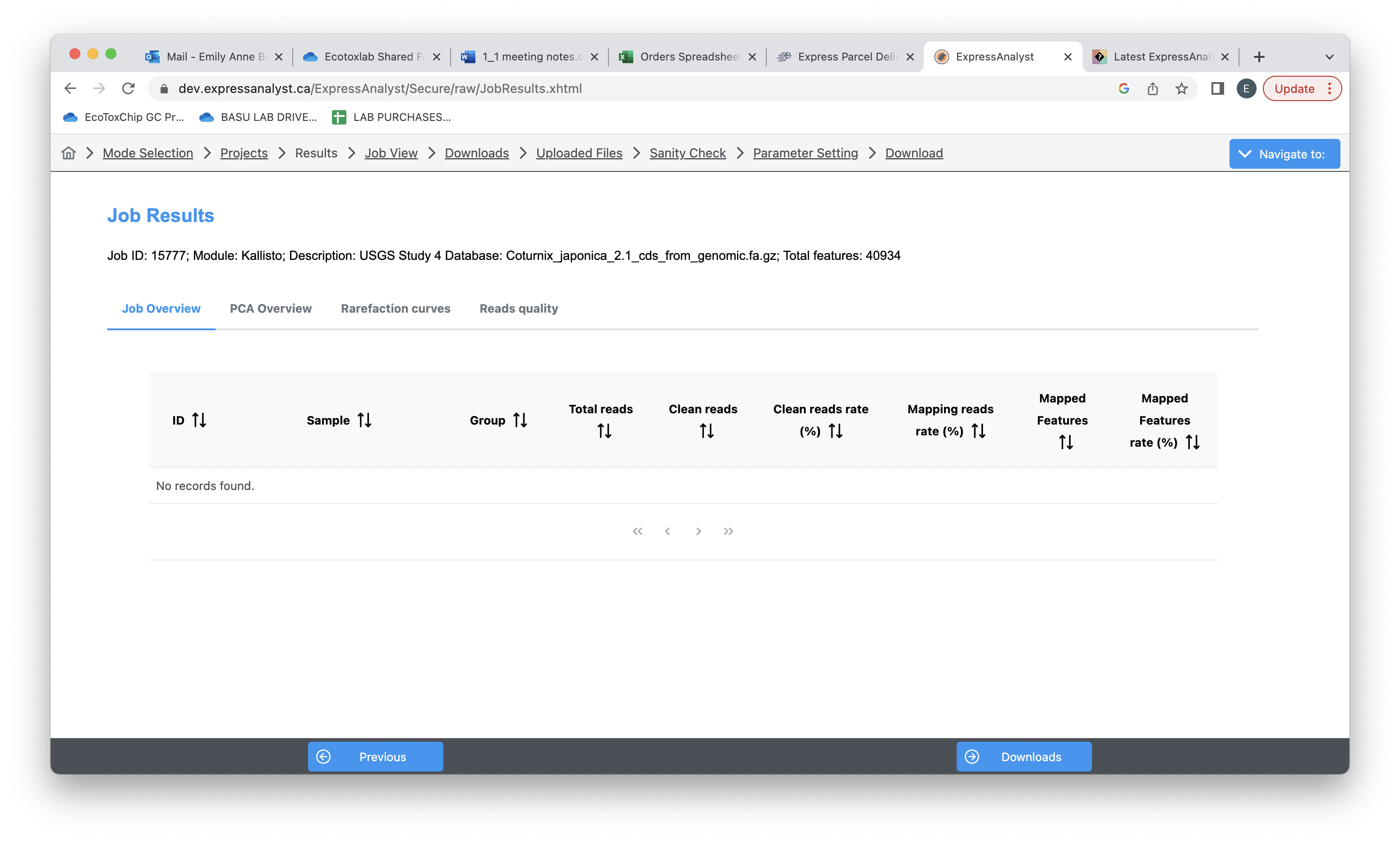
I tried processing single-end FASTQ files (with Japanese Quail Reference - Kallisto) but I don’t get any data…
I’m not sure if I’m doing something wrong, as I’ve never processed single-end files before. I was using a dataset that’s already on ExpressAnalyst (under Reference → USGS Study 4). Please find attached the images of my workflow.
I consulted with Peng and it seems like for single ended RNA-seq data you need to specify these two values for single-ended data: Fragment length and SD of fragment length. We tried with 350 and 50 respectively and we are able to get the results.