Hi teams!
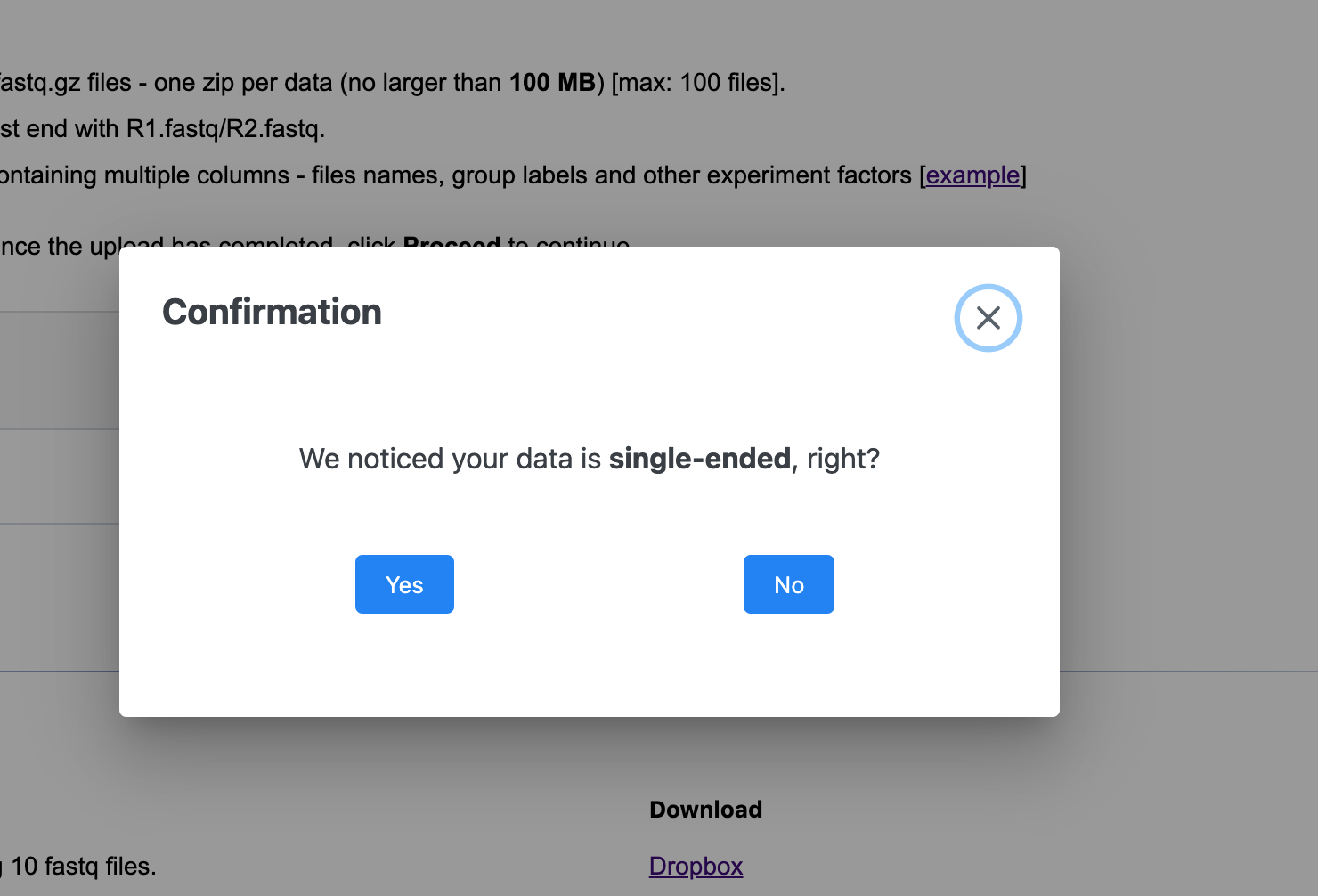
I encountered a difficulty when I provided some paired-end rawdata. After I uploaded the data for processing, the system has been prompting me that “the data you provided is single-ended sequencing, right?” However, it is clear that mine is paired-ended rawdata. Below I attached my metadata
data. I hope to get your help as soon as possible. Thank you.
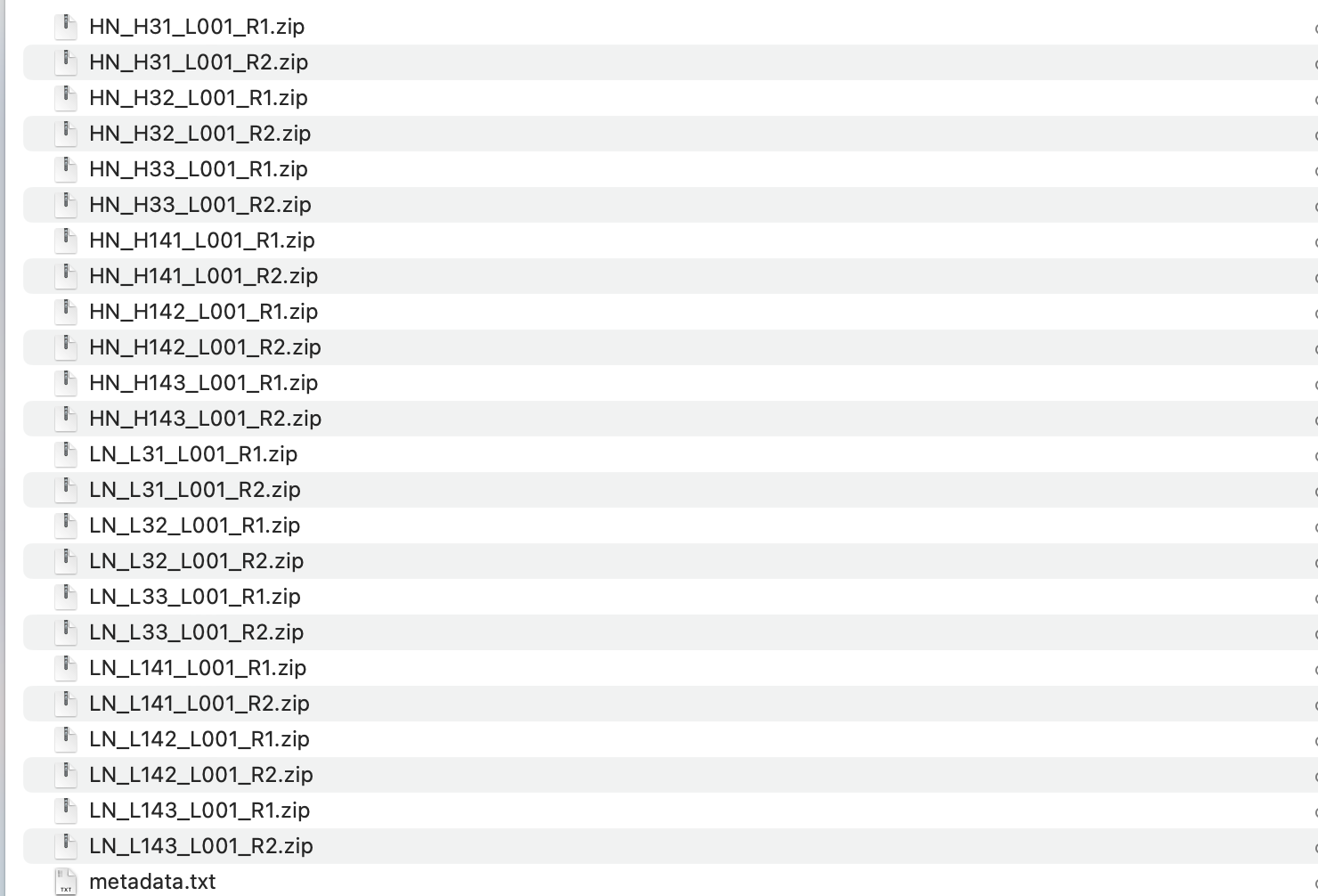
metadata.txt (698 Bytes)
Hello ,
Could you please double check the file names before zipping them. They should end with _R1.fastq/_R2.fastq The paired samples should have the exactly same name except the postfix.
Hope this helps! Please let us know if you have more questions.