Hi,
I have a proteomics dataset I am trying to normalize using Express Analyst but I am returning about 5% unmatched features because it is converting mouse genes. Additionally my dataset has some peptides that map to a group of genes; i.e. [Arxes1;Arxes2], or [Pcdha10;Pcdha4;Pcdha7;Pcdha9] etc. These groups still have relevant information, but it seems they are being filtered out because they are not mapping. I also have two rows of concentrations for some genes, like two separate peptides for Mbp, and I am unsure how to annotate them so they will be included as well.
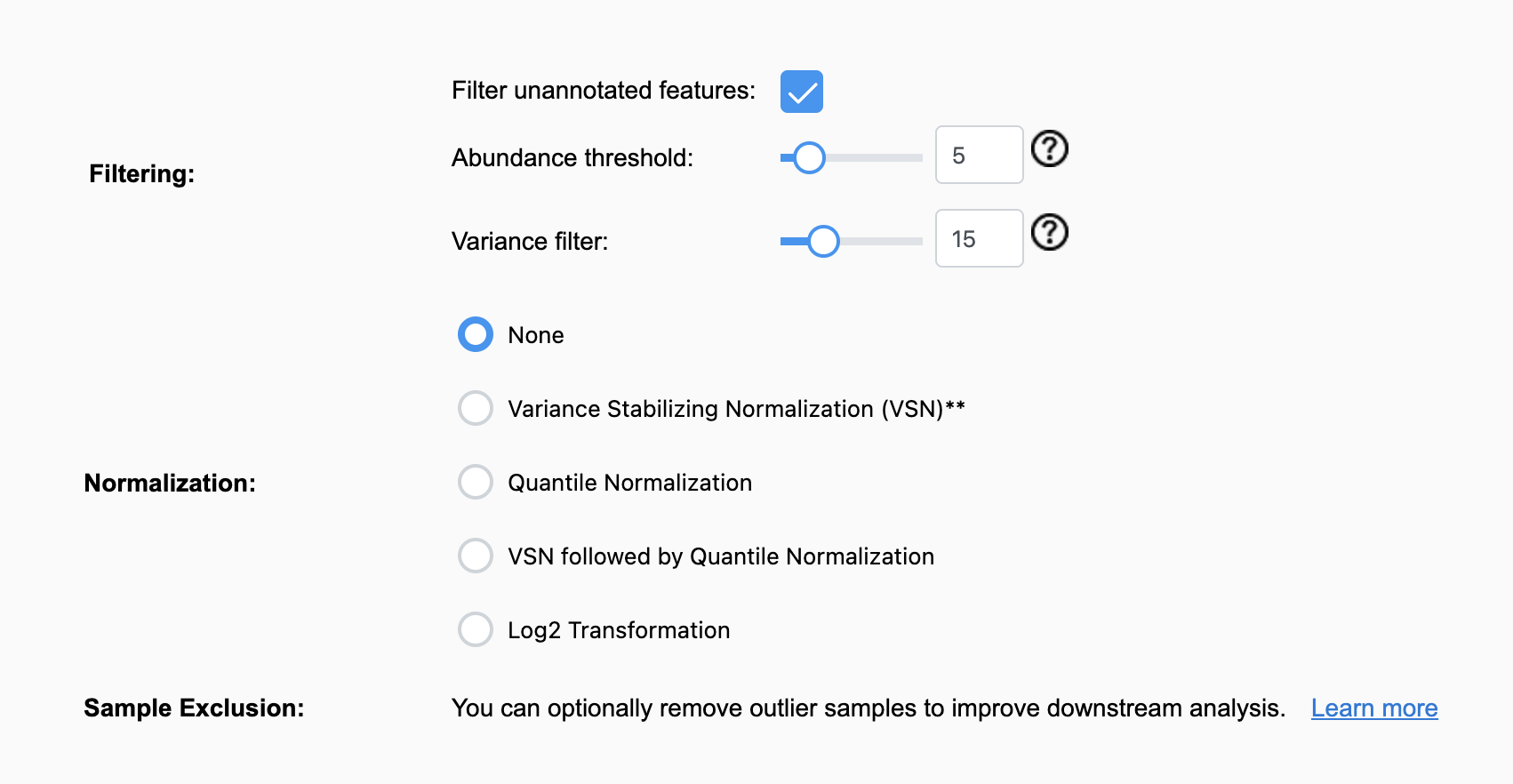
It seems these features that do not map are not included in the normalization process and cannot be circumvented in the web tool? Is it possible in ExpressAnalyst R to ignore the matched vs unmatched features so they can all be included in the normalization and Fold change analysis? I understand the unmapped features being irrelevant for the downstream analysis here, but I want to use the normalized dataset in the MixOmics Pipeline and the full FC list in other services like DAVID and WebGestalt for cross comparison.
Any tips about this would be helpful.
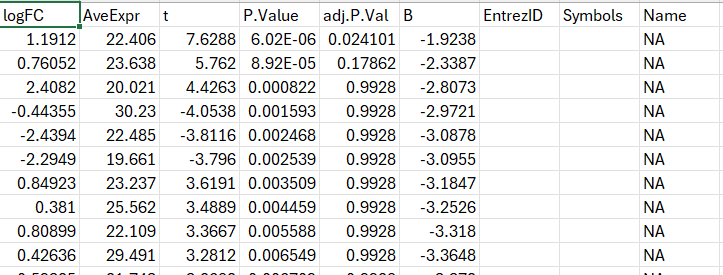
I tried uploading the dataset with ID type --Not Specified --, but the returned FC dataset did not return my input list and instead left the columns blank :