In differential expression analysis, you should first determine whether any of the metadata encode blocking factors, then decide on how to classify individual samples into groups, and finally decide which groups of samples should be compared to each other using statistical tests.
Let’s assume that none of your metadata are blocking factors (more on that later) and try to understand how selecting primary and secondary factors creates different groups of samples. Consider the “Estrogen” example data, generated in a study that measured gene expression at multiple time points in breast cancer cells in which the estrogen receptor (ER) was either present or absent. Here, the metadata are “ER” and “TIME”. As the figure below shows, selecting “ER” as the primary factor divides the data into two groups because “ER” has two different levels (‘present’ and ‘absent’). Selecting “TIME” as the secondary factor results in four groups because the two primary groups are split based on the two time points. If there were three time points, each primary group would be split into three groups, resulting in six groups overall.
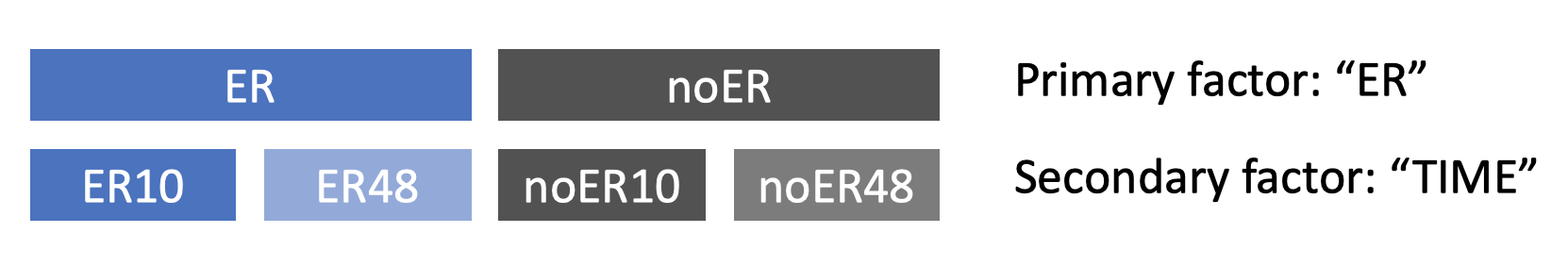
The defined groups can now be compared to find genes that are differentially expressed between them. In some experimental designs, we aren’t interested in finding the genes that are differentially expressed between the groups defined by the secondary factor because it is a blocking factor. Examples of blocking factors are subject IDs when multiple samples were taken from the same subject (e.g. paired samples, multiple tissue types), or batches of samples that were measured at different times or in different locations. If you indicate that your secondary factor is a blocking factor, ExpressAnalyst will conduct comparisons within the groups that it defines, which could potentially improves the accuracy of the overall result.