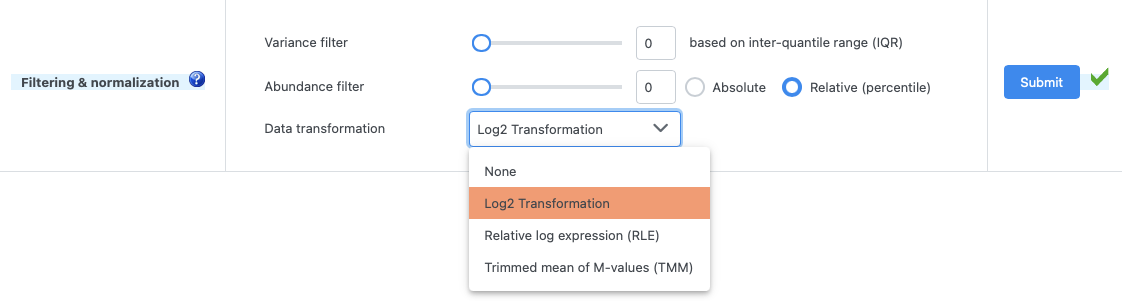
I’m trying to use the “Multiple expression tables” tool in order to complete a meta-analysis for RNA-seq datasets. I’m inputting raw gene counts from htseq-count (not normalized).
According to this paper (https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/cpz1.922), all tools under the normalization drop down menu should normalize your data for sequencing depth, and I just wanted to confirm it was the case for the “log2 transformation” option and that it was not just transforming fold changes into log2FC.